
Avelumab (Bavencio) is a PD-L1 inhibitor that was approved for the treatment of patients with metastatic Merkel cell carcinoma (MCC).
Approval was based on data from an open-label, single-arm, multi-center clinical trial (JAVELIN Merkel 200 trial) demonstrating a clinically meaningful and durable overall response rate (ORR). All patients had histologically confirmed metastatic MCC with disease progression on or after chemotherapy administered for metastatic disease.
ORR was assessed by an independent review committee according to Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. The ORR was 33% (95% confidence interval [CI]: 23.3, 43.8), with 11% complete and 22% partial response rates. Among the 29 responding patients, the response duration ranged from 2.8 to 23.3+ months with 86% of responses durable for 6 months or longer. Responses were observed in patients regardless of PD-L1 tumor expression or presence of Merkel cell polyomavirus.
Safety data were evaluated in 1738 patients who received avelumab, 10 mg/kg, every 2 weeks. The most common serious adverse reactions to avelumab are immune-mediated adverse reactions (pneumonitis, colitis, hepatitis, adrenal insufficiency, hypo- and hyperthyroidism, diabetes mellitus, and nephritis) and life-threatening infusion reactions. Among the 88 patients enrolled in the JAVELIN Merkel 200 trial, the most common adverse reactions were fatigue, musculoskeletal pain, diarrhea, nausea, infusion-related reaction, rash, decreased appetite, and peripheral edema. Serious adverse reactions that occurred in more than one patient in the trial were acute kidney injury, anemia, abdominal pain, ileus, asthenia, and cellulitis.
What is Merkel cell carcinoma?
Merkel cells are neuroendocrine cells in the skin that transmit touch sensitivity to the nervous system. They are located in the epidermis, however, invade into the dermis and deeper structures and blood vessels when becoming cancerous.

Figure 1. Cross-section of skin, normal Merkel cells are shown in red and connect to nerves shown in yellow. The structures drawn include the epidermis (upper third), dermis (middle), and deeper adipose (fatty) layer. Arteries are depicted as red and veins are blue. Diagram of normal Merkel cells in the skin. Figure Copyright by Paul Nghiem, MD, PhD & Quade Medical Group. https://merkelcell.org/about-mcc/what-is-a-merkel-cell/
MCC is caused by infection with the Merkel cell polyomavirus (80%) and exposure to sunlight (20%). Those arising in sun-protected areas of the body are more likely to be associated with an infectious etiology (see below). Two-thirds of patients diagnosed with MCC are over the age of 70.

Figure 2. MCCs are more likely to arise on the left side, likely in part because of greater UV-exposure to the left side when driving. https://merkelcell.org/resources/pictures-of-merkel-cell-carcinoma/

Figure 3. This MCC contained the Merkel cell polyomavirus. MCCs in sun-protected areas are more likely to be driven by the virus than MCCs arising on sun-exposed skin. https://merkelcell.org/resources/pictures-of-merkel-cell-carcinoma/
Chemotherapy with etoposide (a topoisomerase II inhibitor that blocks DNA replication) and carboplatin (an agent that induces inter- and intra-strand cross-links leading to replication arrest and apoptosis) is the treatment of choice for metastatic MCC. Unfortunately, this regimen is not very effective – in a study of 62 patients, 60% experienced tumor shrinkage, however, by day 90, tumors began to grow again.
How does MCC develop?
MCC is strongly linked with infection of a human polyomavirus, Merkel Cell Polyomavirus (MCPyV or MCV). The large T antigen of MCPyV contains Merkel cell specific oncogenic mutations.

Figure 4. A) Schematic of the MCPyV prototype MCV350. The double-stranded DNA genome is approximately 5.4 kilobases in length. The bi-directional origin of replication (white) promotes temporal expression of discrete regions of the genome. The early region encodes three viral T antigens: large T antigen (LT; red), 57 kT antigen (57 kT; orange) and small t antigen (sT; yellow). The late region is transcribed in the opposite direction from the origin and encodes the viral structural proteins: the major capsid protein Virus Protein 1 (VP1; green), and the minor capsid proteins Virus Protein 2 and Virus Protein 3 (VP2; blue, VP3; gray). B) Transmission electron micrograph of MCPyV virus-like particles composed of VP1 and VP2 proteins. The VP1 and VP2 proteins self-assemble into ~50 nm icosahedral particles. (Scale bar=100 nm). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3522868/figure/F2/
A common characteristic of viral oncogenesis is a combination of events that renders the virus incapable of replication, yet still proficient in the induction of cellular proliferation. The LT antigen sequences in almost all tumor-derived MCPyV isolates and MCC cell lines analyzed to date contain mutations within exon 2 that prematurely truncate the LT protein. Strikingly, these mutations all occur within the carboxy terminus of LT at sites downstream of the LXCXE Rb-binding motif. This consistent, tumor-specific mutational pattern gives rise to truncated LT antigens in which the CR1, DnaJ, MUR, NLS, and pRb-binding motifs are preserved, yet domains associated with replication are lost. Indeed, truncated LT proteins can still co-immunoprecipitate pRb but are defective for origin-binding and viral helicase activities, and are thus no longer functional in replication assays. These mutations in LT lie within a region that does not affect the coding capacity for sT. The presence of tumor-specific mutations within the MCPyV genome is compelling evidence that the virus has lost the capacity to replicate in MCC tumors, but retains motifs with potential to contribute to uncontrolled proliferation.
Binding of mutated MCPyV’s Large T antigen to Rb occupies the pocket in which E2F transcription factors bind to Rb, thereby liberating them to usher uncontrolled cellular proliferation. The mutated protein also binds to a neutralizes p53.

Figure 5. Mutated MCPyV’s Large T antigen antigen binds to Rp and p53 – it no longer induces lysis, but it does induce tumor transformation. https://www.slideshare.net/guesta37aeb/mcpyv-presentation-presentation/33
Another checkpoint inhibitor that disrupts the PD-1/PD-L1 axis
Avelumab is the fourth monoclonal antibody that disrupts the PD-1/PD-L1 axis that has been approved by the FDA – nivolumab (Opdivo, PD-1: melanoma, squamous cell carcinoma of the head and neck – HNSCC, non-small cell lung cancer – NSCLC, bladder cancer, renal cell carcinoma, classical Hodgkin lymphoma), pembrolizumab (Keytruda, PD-1: NSCLC, HNSCC, classical Hodgkin Lymphoma), and atrezolizumab (Tecentriq, PD-L1: non-small cell lung cancer and bladder cancer). A fifth antibody, darvalumab (PD-L1), is in development for non-small cell lung cancer, head and neck, bladder, gastric, pancreatic, hepatocellular carcinoma, and blood cancers.
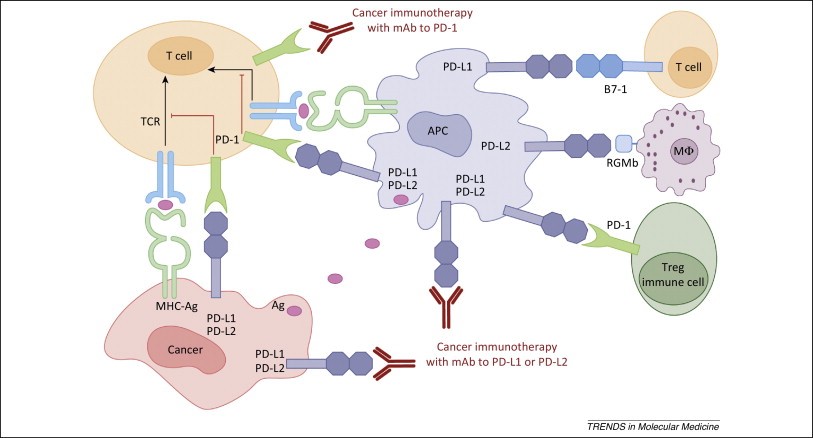
Figure 6. Human cancer immunotherapy with anti-PD-1 and anti-PD-L1/L2 antibodies. Antigen-presenting cells (APC) take up antigens (Ag) released from cancer cells and present to T cells. Cancer cells can also present Ag to activated T cells in the context of MHC. Upon T cell activation, PD-1 receptors are expressed on T cells and inhibit immune responses by engagement of PD-L1 and PD-L2 ligands on APC and PD-L1 on cancer cells. Therefore, monoclonal antibody (mAb)-mediated specific blockade of the PD-1/PD-L1/PD-L2 pathway can enhance anti-tumor immunity. In addition to binding to PD-1, PD-L1 and PD-L2 also bind B7-1 and repulsive guidance molecule B, respectively. In addition to T cells and APC, PD-1 and PD-L1 can be induced on other immune cells. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4282825/