Just when we thought we understood the cell biology of antimicrobial resistance in macrophages, a study like the one detailed by Abuaita and colleagues in their Cell Host & Microbe article comes along.[1] Not only are the molecular mechanisms of pathogen metabolism updated, but new cellular signaling and trafficking pathways are implicated. There is a lot of new and important science included in this exciting paper.
Let’s begin by remembering that macrophages are immune cells whose role is to internalize and degrade invading microorganisms. They do so by endocytosing the pathogenic microbes and sequestering them in a newly developed phagosome. It is there that the destructive processes are initiated; final elimination of residual components occurs in the lysosome – a related degradative organelle that works in close concert with the nascent phagosome.
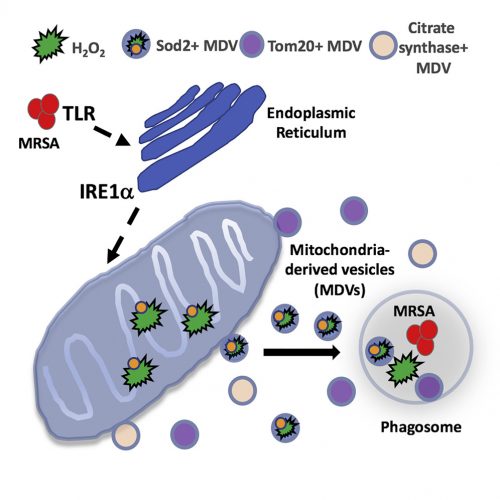
What Abuaita and co-authors describe is a process by which certain bacteria, including the potentially dangerous methicillin-resistant Staphylococcus aureus, trigger more than simply a phagocytic event. Rather, it is clear that the phagosome somehow recognizes the microorganism within its midst, and launches a far more lethal and complex biochemical reaction. The cascade begins with the infected macrophage mounting a stress response – one initiated by the phagosome but further elaborated by the endoplasmic reticulum. Specifically, the endoplasmic reticulum turns on one of the most elegantly choreographed and well-described quality-control systems in the cell, called the unfolded protein response, or UPR. The critical sensor that needs to be tripped to turn on the pathway is the endoplasmic reticulum membrane protein, IRE1a. It is at this point that the cell biology really gets exciting.
The response is not confined to the endoplasmic reticulum and well understood downstream nuclear effector pathways; rather, the signal is somehow transmitted to mitochondria. In response, the organelle is prompted to produce reactive oxygen species – hydrogen peroxide in particular. Mitochondria are known to produce reactive oxygen species, including hydrogen peroxide, as part of their normal energy-generating metabolism. What is different here is that a defense mechanism initiated in the phagosome, signals the endoplasmic reticulum, which then communicates to the mitochondria; a truly elegant and previously unrecognized relay system.
As interesting as the new interorganellar signaling events are – there is more. Specifically, the fascinatingly novel trafficking pathways identified.
The newly synthesized mitochondrial hydrogen peroxide is packaged in vesicles which are shed from the organelle. The authors even identify a critical component of this release step – the ubiquitin ligase, Parkin. These newly created, hydrogen peroxide-loaded mitochondrial vesicles then migrate through the cell, destined to fuse with pathogen-infected phagosomes – releasing their toxic contents and supplementing the already initiated bacterial killing process. Just in case the newly delivered hydrogen peroxide does not provide a sufficient amount of toxic reactive oxygen, some mitochondrial vesicles actually encapsulate the hydrogen peroxide-synthesizing enzyme, superoxide dismutase-2.
What we have here is the cell marshalling its degradative armamentarium in a manner we never imagined to help fight methicillin-resistant Staphylococcus aureus and other invading pathogens. It seems clear that this is only the tip of the iceberg – interorganelle communication/signaling networks and elaborately coordinated effector apparatuses in cells are certain to exist in ways we can only begin to imagine. In this case, the goal is to fight infection; in others, it may very well be to thwart the effects of aging, to counter disease, or neutralize the effects of mutations, toxins, or other environmental insults. The more we know about cell biology, the more we realize how much we do not know; it is amazing how the field continues to captivate our attention.
SRT – February 2019
[1]B.H. Abuaita et al., Cell Host and Microbe (2018) doi: 10.1016/j.chom.2018.10.005. PMID: 30449314 (Request article via ILL).