
Overview of a Literature Review
A literature review is a generic term used to describe a synthesis of information to answer a research question. The purpose of a literature review is to present the scholarly information that is available on a topic, provide support to the proposed research, and relate the literature to the proposed research question. There are numerous types of literature reviews. These vary from a narrative review to a systematic review.
Review types differ by:
-
-
- the precision of the research question (broad to specific)
- the goal of the review
- the standards of the searching method
- if the articles are appraised
- how information from various sources is synthesized
- the analysis of the results
- showing the current state of the literature around a particular topic
-
The IHS Library offers assistance “How to Write a Literature Review.”
Types of Literature Reviews
Literature or Narrative Review
-
- Team: May be completed by a single author
- Definition: Generic term: A synthesis of current literature surrounding a specific topic. The purpose of a narrative review is to provide background information on the topic, support the proposed research and/or answer a research question.
- Search Methods: Non-specific; Author chooses relevant articles based on research question.
- Appraisal: Determined by the author
- Synthesis: Narrative
- Analysis: Chronological, conceptual, thematic, etc.
Scoping or Mapping Review
-
- Team: Requires a minimum of 2 authors
- Definition: Preliminary assessment of potential size and scope of available research literature on a broad topic. Aims to identify nature and extent of research evidence. Includes grey literature, preprints and ongoing studies. Scoping reviews are conducted based upon the JBI manual of evidence synthesis.
- Search Methods: Broad scope of literature available. Search methods must be transparent and reproducible. Search strategies are peer reviewed & documented in full.
- Appraisal: All evidence is independently screened by 2 reviewers to ensure evidence meets the inclusion criteria. The critical appraisal process is optional but recommended
- Synthesis: Narrative
- Analysis: Characterizes quantity and quality of literature based upon the elements of the PCC research question and the inclusion/exclusion criteria
Systematic Review
-
- Team: (Requires a minimum of 2 authors)
- Definition: Seeks to systematically search for, appraise and synthesize all available research evidence on the topic. SRs answer a specific research question and are conducted based upon the JBI manual of evidence synthesis.
- Search Methods: Exhaustive, comprehensive, & systematic search. Search methods must be transparent & reproducible. Search strategies are peer reviewed & well documented.
- Appraisal: All evidence is independently screened by 2 reviewers to meet inclusion criteria and critically appraised using the JBI Critical Appraisal Checklists
- Synthesis: Narrative
- Analysis: Synthesizes what is known within the existing literature. Highlights what is unknown and recommends future research.
- Umbrella Review
-
- Team: (Requires a minimum of 2 authors)
- Definition: Reviews the results of multiple systematic reviews on a specific topic. All reviews must analyze a shared methodology, facilitating comparison and analysis. Umbrella reviews are conducted based upon the JBI manual of evidence synthesis
- Search Methods: Exhaustive, comprehensive & systematic search of reviews. Does not include primary studies. Search methods must be transparent, reproducible, and well documented.
- Appraisal: All evidence is independently screened by 2 reviewers to meet inclusion criteria and critically appraised using the JBI Critical Appraisal Checklists
- Synthesis: Graphical and tabular with narrative commentary
- Analysis: What is known; Recommendations for practice. What remains unknown; recommendations for future research
Rapid Review
-
- Team: Requires a minimum of 2 authors
- Definition: Assessment of what is already known about a policy or practice issue, by using systematic review methods to search and critically appraise existing research. RRs are conducted according to the JBI manual of evidence synthesis
- Search Methods: Completeness of searching determined by time constraints. All search strategies must be transparent, reproducible, and documented
- Appraisal: Time-limited formal quality assessment.
- All evidence is independently screened by 2 reviewers to meet inclusion criteria
- Synthesis: Narrative and tabular
- Analysis: Quantities of literature and overall quality/direction of effect of literature
Meta Analysis
-
- Definition: Statistical analysis of quantitative evidence provided within a Systematic Review.
- Team: Interdisciplinary
- Meta-analysis are conducted according to the JBI manual of evidence synthesis
- Search Methods: Exhaustive, comprehensive & systematic search of reviews. Does not include primary studies. Search methods must be transparent, reproducible and documented.
- Appraisal: All evidence has been critically appraised in the systematic review
- Synthesis: Graphical representation in a Forest plot.
- Analysis: Numerical analysis of measures of effect assuming absence of heterogeneity
Reproduced from Grant, M. J. and Booth, A. (2009), A typology of reviews: an analysis of 14 review types and associated methodologies. Health Information & Libraries Journal, 26: 91–108. doi:10.1111/j.1471-1842.2009.00848.x
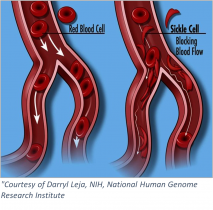 Sickle cell anemia is an inherited blood-borne disease affecting some 100,000 individuals in the US, and nearly 8 million worldwide. Persons of sub-Saharan African descent appear to manifest the disease to the greatest extent, with those of Indian, Hispanic, or Middle Eastern backgrounds also highly affected. The pathology is devastating – misshapen red blood cells occlude blood vessels, compromising flow and inhibiting oxygen delivery. Pain develops frequently in oxygen-deprived tissues. Other complications include an enhanced susceptibility to infections, various eye problems, organ damage, and increased risks of pulmonary/heart disease and stroke.
Sickle cell anemia is an inherited blood-borne disease affecting some 100,000 individuals in the US, and nearly 8 million worldwide. Persons of sub-Saharan African descent appear to manifest the disease to the greatest extent, with those of Indian, Hispanic, or Middle Eastern backgrounds also highly affected. The pathology is devastating – misshapen red blood cells occlude blood vessels, compromising flow and inhibiting oxygen delivery. Pain develops frequently in oxygen-deprived tissues. Other complications include an enhanced susceptibility to infections, various eye problems, organ damage, and increased risks of pulmonary/heart disease and stroke.