
Terlecky’s Corner: Installment 12
Gene Therapy for Sickle Cell Anemia: Repairing Hemoglobin Subunit Assembly
This month’s announcement[1] from the Food and Drug Administration (FDA) that it will approve two therapeutic approaches to address the molecular defects associated with sickle cell anemia is a major step forward not only in terms of treatment of the disease, but also as evidence of how far the science of gene editing has come.
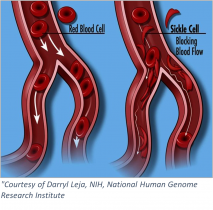 Sickle cell anemia is an inherited blood-borne disease affecting some 100,000 individuals in the US, and nearly 8 million worldwide. Persons of sub-Saharan African descent appear to manifest the disease to the greatest extent, with those of Indian, Hispanic, or Middle Eastern backgrounds also highly affected. The pathology is devastating – misshapen red blood cells occlude blood vessels, compromising flow and inhibiting oxygen delivery. Pain develops frequently in oxygen-deprived tissues. Other complications include an enhanced susceptibility to infections, various eye problems, organ damage, and increased risks of pulmonary/heart disease and stroke.
Sickle cell anemia is an inherited blood-borne disease affecting some 100,000 individuals in the US, and nearly 8 million worldwide. Persons of sub-Saharan African descent appear to manifest the disease to the greatest extent, with those of Indian, Hispanic, or Middle Eastern backgrounds also highly affected. The pathology is devastating – misshapen red blood cells occlude blood vessels, compromising flow and inhibiting oxygen delivery. Pain develops frequently in oxygen-deprived tissues. Other complications include an enhanced susceptibility to infections, various eye problems, organ damage, and increased risks of pulmonary/heart disease and stroke.
At the molecular level – sickle cell anemia (in its most common form) is the result of a faulty hemoglobin protein inside red blood cells. Hemoglobin – the vehicle for oxygen delivery in our bodies – is composed of four subunits, two alpha- and two beta-globin proteins, with each complexing an iron-containing heme prosthetic group. Hemoglobin is a marvel of protein biochemistry – a paradigm for allosteric (in this case, oxygen-binding) cooperativity. That is, when the first oxygen molecule binds to hemoglobin, binding of the second oxygen is “cooperatively” enhanced; and similarly for oxygen additions three and four. The fully loaded hemoglobin then leaves the lungs and travels through the blood to tissues whereupon oxygen is released. This is the normal circumstance.
In sickle cell anemia, amino acid 6 of the beta-globin subunit is altered – from glutamic acid to valine. Any protein biochemist will readily recognize that such a substitution (charged reside to hydrophobic one) could dramatically change the molecules folding and/or functional properties. Such is the case with the beta-globin protein – which now interacts inappropriately with other (beta-globin) subunits by virtue of the newly exposed (hydrophobic) surface. As a result, hemoglobin’s tertiary (that is, folded) structure is altered. Indeed, the misshapen hemoglobin molecule aberrantly polymerizes and forms long fibers with the resultant deformed (~sickle shaped) red blood cells causing the aforementioned vaso-occlusive manifestations.
To best understand the genetic strategies employed in the newly approved therapies, some mention of fetal hemoglobin is warranted. Fetal hemoglobin, like the adult version, is a tetrameric protein – with two alpha-subunits – which are complexed with two gamma-, not beta-globin subunits. Shortly after birth, a switch occurs – gamma-globin synthesis is reduced and beta-globin’s turned on. Beta-globin now replaces gamma-globin in complexing with alpha-globin chains to create the adult hemoglobin molecule.
Interestingly, some patients with sickle cell disease continue to make fetal hemoglobin, and enjoy a milder disease course. In fact, the previously FDA-approved drug hydroxyurea, which helps boost fetal hemoglobin levels, has shown efficacy in treating the disease. (On the down side, concerns about toxicity and uneven effectiveness across patient populations have limited hyroxyurea’s more universal adoption.) Nevertheless, the anti-sickling properties of fetal hemoglobin’s gamma-globin chain have been recognized.
Scientists compared gamma-globin to beta-globin and tested variously altered beta-globin derivatives that would confer gamma-globin’s anti-sickling property. One such alteration is a threonine to glutamine change at position 76. It is lentivirus-mediated expression of beta-globin(T76Q) into hematopoietic stems cells that constitutes the basis of Bluebird Bio’s Lyfgenia® (lovotibeglogene autotemcel) therapy – approved by the FDA on December 8th. The idea is that the non-sickling beta-globin(T76Q) subunits will complex with alpha-subunits (and heme prosthetic groups) and result in a fully functional hemoglobin molecule.
Casgevy® (exagamglogene autotemcel) from Vertex Pharmaceuticals takes a different approach. It employs the CRISPR/Cas9 (gene editing) system to eliminate production of a protein called B-cell lymphoma/leukemia 11A (BCL11A). BCL11A is an enzyme which induces the switch in humans – shifting expression from gamma-globin to beta-globin. As described above, this occurs during human development – specifically at birth. As the strategy will result in (non-sickling) gamma-globin production – once again functional hemoglobin will be produced.
To varying degrees, both strategies appear to work – hence the FDA’s approval. There is risk – as the intricate therapeutic approaches require: i. removal of hematopoietic stem cells; ii. the genetic alterations as outlined; iii. conditioning of the patient for receipt of the genetically engineered replacement cells; and iv. the cells’ reintroduction. Also, as might be expected from the complex nature of the steps involved, even as one-time therapies, they are both extremely expensive. That said, how can a price be placed on enjoying even modest relief from the pain and suffering associated with sickle-cell disease?
SRT – December 2023
[1] https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease