
We have discussed mutational burden previously on this blog – in essence, the concept is that tumors with more mutations are more visible to the immune system because the generation of new novel antigenic epitopes allows for adaptive immune responses even when previous adaptive antigen-specific immune responses have been blunted by PD-1 expression.
It follows that anti-PD1 therapy would work best against cancers that present neo-antigens to the immune system since these have a higher likelihood of expressing PD-1 and a lower likelihood of having induced antigen-specific Treg’s. The more immunogenic the tumor, the more likely that anti-PD1 therapy will be effective since PD-1 is an immune abrogating signal that is induced by tumors with a microenvironment characterized by an IFN-γ inflammatory signature on gene expression profiling (GEP) analysis.
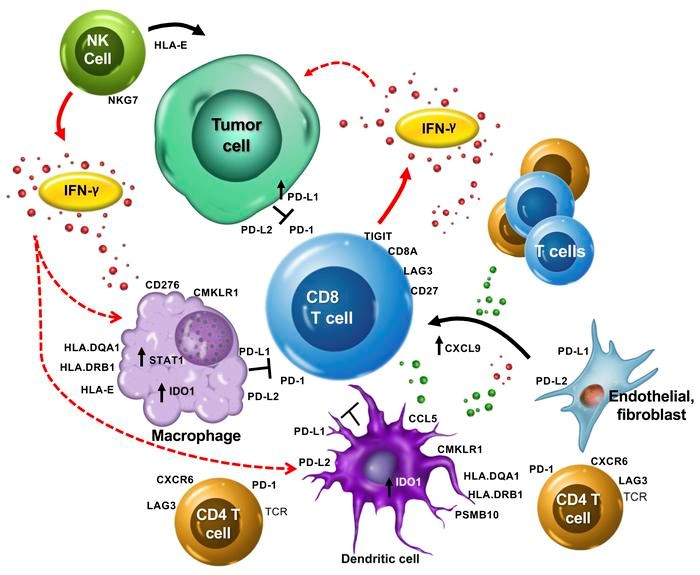
Figure 1. The T cell-inflamed gene expression signature highlights the complex biology of the host immune microenvironment. The production of IFN-γ by activated T cells, and perhaps other cells such as NK and NK T cells, activates a host of key downstream signaling molecules such as STAT1 and CMKLR1 in dendritic cells and macrophages. These cells in turn produce chemokines/chemoattractants, including CCL5 and CXCL9, which recruit additional CD8+ T cells. T cells upregulate activation-induced costimulatory receptors such as CD27 and produce an array of effector molecules, including IFN-γ, perforin, and granzymes. IFN-γ can induce upregulation of HLA molecules and other components of the immunoproteosome and antigen-presenting machinery such as PSMB10. However, IFN-γ also upregulates a host of checkpoint inhibitors, such as PD-L1 and PD-L2, on the surface of macrophages and dendritic and tumor cells. Other checkpoint molecules, such as IDO1, TIGIT, and LAG3, might also be homeostatically upregulated by T cell activation and IFN-γ signaling, and cooperate with PD-1 to restrain the antitumor immune response. The association of genes such as HLA-E and NKG7 with clinical response suggests that additional aspects of NK cell biology or noncanonical T cell antigen recognition might be involved in the T cell–inflamed microenvironment. Overall, the genes identified suggest that multiple immune cell types contribute to a T cell–inflamed but immune checkpoint–restrained tumor microenvironment that can be successfully modulated by PD-1 checkpoint blockade. https://www.jci.org/articles/view/91190
Merck’s Keytruda (pembrolizumab) garnered FDA approval for the treatment of tumors with high microsatellite instability (MSI-H, or mismatch repair deficient – dMMR), irrespective of tissue of origin. This marks the first approval based on a genetic signature, as opposed to cancer type. It was based on analyses from five clinical trials including 149 patients.
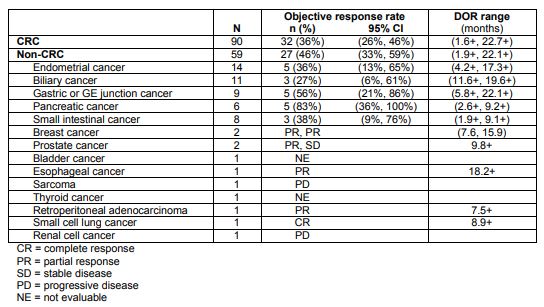
Table 1. Pembrolizumab response in tumors with microsatellite instability-high. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf
MSI-H/dMMR is identified using polymerase chain reaction for mutated mismatch repair genes (MLH1, MSH2, MSH6, or PMS2 ) or immunohistochemistry for loss of expression of these proteins.
Tumor mutational burden (TMB) evaluates to coding sequences of the genome, not the integrity and expression of mismatch repair enzymes. It is evaluated via whole genome sequencing or through targeted sequencing panels – it measures the number of non-synonymous DNA coding sequence changes per megabase of sequenced DNA. (Non-synonymous mutations are base substitutions that result in altered amino acid sequences compared to wild type.)
Researchers at Johns Hopkins looked at 27 different cancer types to evaluate TMB as a as prognostic biomarker for checkpoint therapy. In general, the higher the TMB, the better response to immune checkpoint blockade:
We observed a significant correlation between the tumor mutational burden and the objective response rate (P<0.001). The correlation coefficient of 0.74 suggests that 55% of the differences in the objective response rate across cancer types may be explained by the tumor mutational burden.
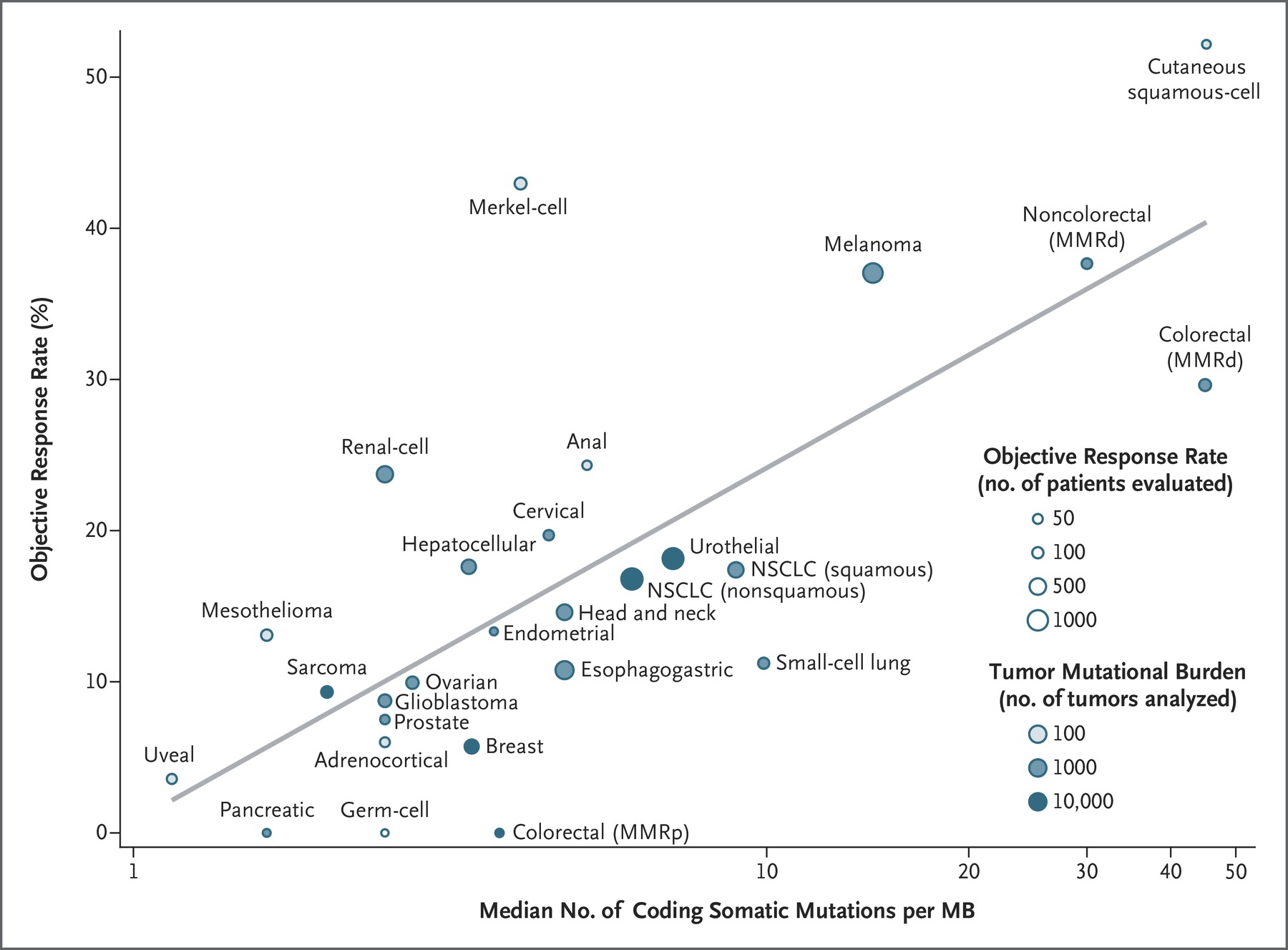
Figure 2. Correlation between Tumor Mutational Burden and Objective Response Rate with Anti–PD1 or Anti–PD-L1 Therapy in 27 Tumor Types. Shown is the median number of coding somatic mutations per megabase (MB) of DNA in 27 tumor types or subtypes among patients who received inhibitors of programmed death 1 (PD-1) protein or its ligand (PD-L1), as described in published studies for which data regarding the objective response rate are available. The number of patients who were evaluated for the objective response rate is shown for each tumor type (size of the circle), along with the number of tumor samples that were analyzed to calculate the tumor mutational burden (degree of shading of the circle). Data on the x axis are shown on a logarithmic scale. MMRd denotes mismatch repair-deficient, MMRp mismatch repair-proficient, and NSCLC non–small-cell lung cancer. https://www.nejm.org/doi/full/10.1056/NEJMc1713444?query=featured_home&
There were some outliers, however:
Some cancer subtypes have a response to therapy that is better than would be predicted by the tumor mutational burden (e.g., Merkel-cell carcinoma), and some have a response that is worse than would be predicted (e.g., colorectal cancer with mismatch repair proficiency). The higher-than-anticipated objective response rates for Merkel-cell carcinoma and some other cancers that have been associated with viruses suggest that the presentation of viral antigens on certain tumor types may confer an increased response rate to anti–PD1 therapy.
As can be seen in Figure 2, Kidney cancer responds well to anti-PD1 therapy, but it does not have a particularly high TMB. This is because kidney cancers have a high number of “indel” (insertion and deletion mutations resulting in frameshifts) mutations, which are highly mutagenic. Colorectal cancers with proficient MMR do not respond as well to anti-PD1 therapy as other tumors with a similar level of TMB. And, tumors driven by oncogenic viruses respond better to anti-PD1 therapy than other tumors of similar TMB because viral proteins are very immunogenic.
“Although mismatch repair deficiency is a biomarker for PD-1 response that already captures some patients with high TMBs, there are clearly other patients with high TMBs who do not have mismatch repair deficiency,” said Mark Yarchoan, MD. “In the future, TMB might guide the use of anti-PD1 therapy in a tissue-agnostic manner, similar to mismatch repair deficiency.”
The researchers are interested in how to get cancers with low mutational burden to be made to behave like those with higher mutational burden: “How do we make immunologically ‘cold’ tumors, which usually have lower TMBs, behave more like immunologically ‘hot’ tumors? We are hopeful that therapeutic cancer vaccines might be able to augment responses to anti-PD1 therapy in ‘cold’ tumors,” said Yarchoan, adding that vaccine combinations may prime the tumor microenvironment to make tumors more amenable to anti-PD1 therapy.