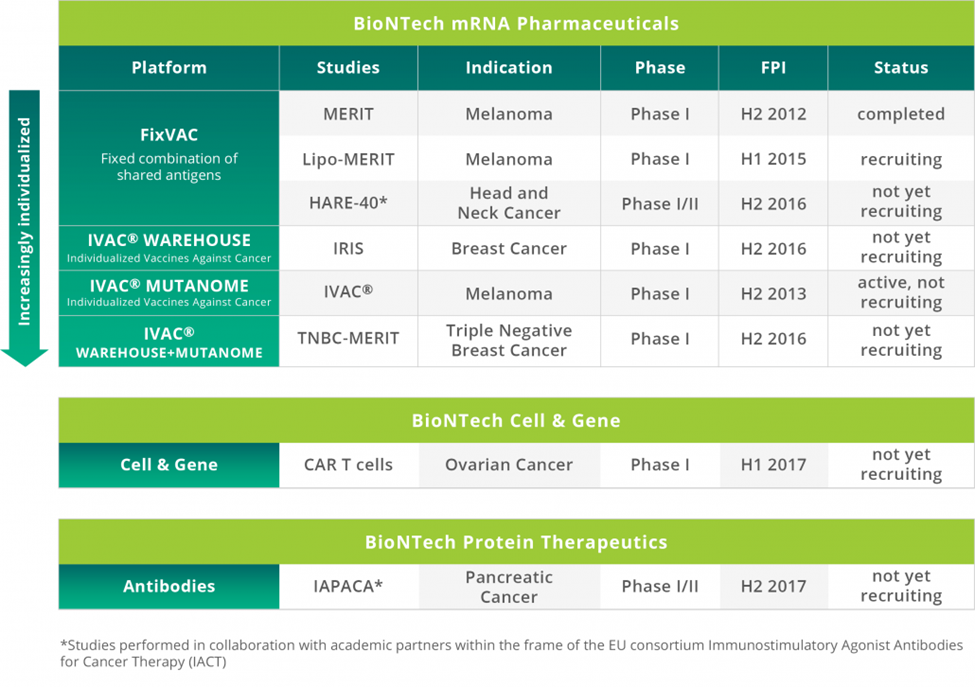
Roche will be collaborating with BioNTech to develop personalized vaccines based on BioNTech’s mRNA products combined with Roche’s PD-L1 checkpoint inhibitor, Tecentriq (atezolizumab), which was approved for bladder cancer in May 2016.
What is atezolizumab?
Atezolizumab belongs to a class of immunotherapy drugs known as checkpoint inhibitors. The drug prevents a protein called PD-L1 that is found on some tumor cells from binding to another protein, PD-1, on immune cells (Figure 1). The binding of these “checkpoint” proteins suppresses the immune response. By blocking this interaction, checkpoint inhibitors “release the brakes” on the immune system, allowing immune cells to attack tumors.
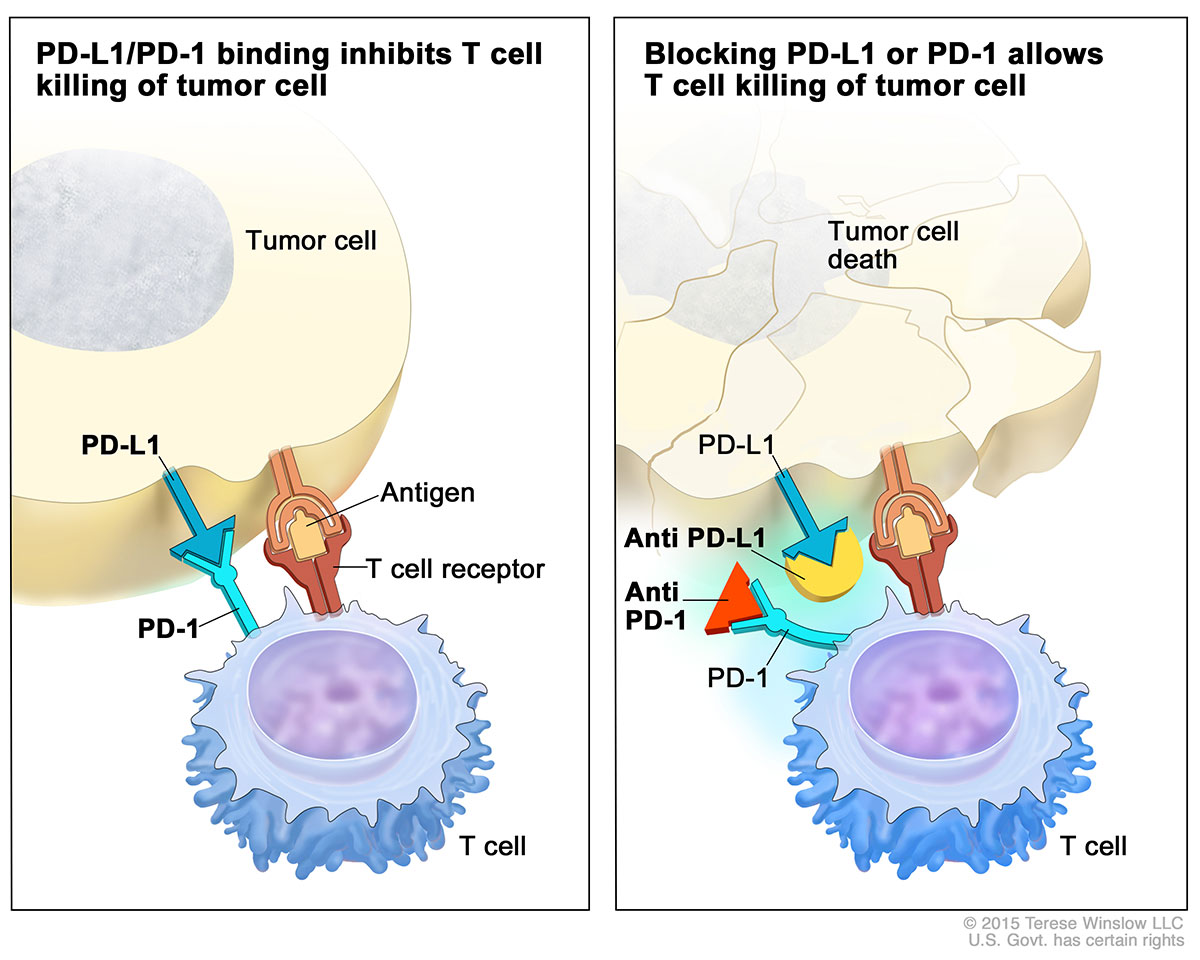
Figure 1 – Atezolizumab blocks the binding on PD-1 receptor on T-cells with PD-L1 ligand on antigen presenting cells, including tumor cells. https://www.cancer.gov/news-events/cancer-currents-blog/2016/fda-atezolizumab-bladder
The approval of atezolizumab was based on a study of 310 patients with locally advanced or metastatic urothelial carcinoma whose cancers had worsened during or after treatment with platinum-containing chemotherapy or within 12 months of receiving platinum-containing chemotherapy, either before or after surgery. All patients in the trial received atezolizumab.
Approximately 15% of patients had at least a partial shrinkage of their tumors, and this effect lasted from at least 2.1 months to more than 13.8 months, the study authors reported.
The most common side effects of treatment with atezolizumab were fatigue, decreased appetite, nausea, urinary tract infection, fever, and constipation. The therapy also may cause infection and serious immune system-related side effects.
In the trial, increased PD-L1 expression in patients’ tumors was associated with response to atezolizumab. Among the trial participants whose tumors were classified as “positive” for PD-L1 expression, 26% experienced a tumor response, compared with 9.5% of the participants whose tumors were “negative” for PD-L1 expression.
At the same time that it approved atezolizumab, the FDA also approved a test, called Ventana PD-L1 (SP142), to measure PD-L1 expression on patients’ tumor-infiltrating immune cells.
What does BioNTech’s mRNA technology?
BioNTech’s approach is to sequence a patient’s tumor genome, then encoding the neoantigens for that particular tumor into a message delivered by mRNA as a vaccine. Dendritic cells decode the information, and use it to mark tumor cells for destruction by the immune system.
Every tumor has an exclusive pattern of shared tumor antigens and mutations that makes every tumor absolutely unique. BioNTech uses next generation sequencing (NGS) and proprietary analytical algorithms to define these tumor-specific antigen signature, or neonantigens, called mutanome. The platform is called Individualized Cancer-immunotherapy (IVAC®, Figure 2).
Castle and colleagues validated the concept of mutanomes and using neoantigens from a mutanome as a vaccine in work performed with B16F10 melanoma:
the Multiple genetic events and subsequent clonal evolution drive carcinogenesis, making disease elimination with single-targeted drugs difficult. The multiplicity of gene mutations derived from clonal heterogeneity therefore represents an ideal setting for multi-epitope tumor vaccination. Here, we used next generation sequencing exome resequencing to identify 962 nonsynonymous somatic point mutations in B16F10 murine melanoma cells, with 563 of those mutations in expressed genes. Potential driver mutations occurred in classical tumor suppressor genes and genes involved in proto-oncogenic signaling pathways that control cell proliferation, adhesion, migration, and apoptosis. Aim1 and Trrap mutations known to be altered in human melanoma were included among those found. The immunogenicity and specificity of 50 validated mutations was determined by immunizing mice with long peptides encoding the mutated epitopes. One-third of these peptides were found to be immunogenic, with 60% in this group eliciting immune responses directed preferentially against the mutated sequence as compared with the wild-type sequence. In tumor transplant models, peptide immunization conferred in vivo tumor control in protective and therapeutic settings, thereby qualifying mutated epitopes that include single amino acid substitutions as effective vaccines. Together, our findings provide a comprehensive picture of the mutanome of B16F10 melanoma which is used widely in immunotherapy studies. In addition, they offer insight into the extent of the immunogenicity of nonsynonymous base substitution mutations. Lastly, they argue that the use of deep sequencing to systematically analyze immunogenicity mutations may pave the way for individualized immunotherapy of cancer patients.
How will mRNA be targeted to dendritic cells?
Key for RNA vaccines to elicit an adaptive immune response is successful dendritic cell targeting and presentation of epitopes encoded by the mRNA on both MHC Class I and MHC Class II molecules (Figure 3). Micropinocytosis of the RNA permits processing within the endosomal pathway, that is, translation of the mRNA.
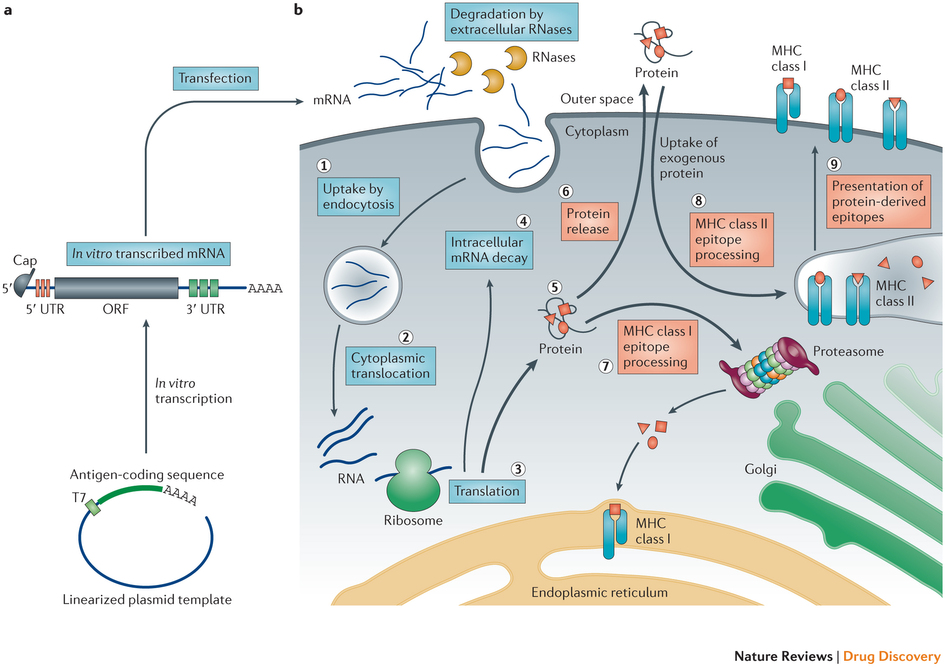
Figure 3 – Principles of antigen-encoding mRNA pharmacology.
a | A linearized DNA plasmid template with the antigen-coding sequence is used for in vitro transcription. The in vitro transcribed mRNA contains the cap, 5′and 3′ untranslated regions (UTRs), the open reading frame (ORF) and the poly(A) tail, which determine the translational activity and stability of the mRNA molecule after its transfer into cells. b | Step 1: a fraction of exogenous mRNA escapes degradation by ubiquitous RNases and is spontaneously endocytosed by cell-specific mechanisms (for example, macropinocytosis in immature dendritic cells) and enters endosomal pathways. Step 2: release mechanisms of mRNA into the cytoplasm are not fully understood. Step 3: translation of mRNA uses the protein synthesis machinery of host cells. The rate-limiting step of mRNA translation is the binding of the eukaryotic translation initiation factor 4E (eIF4E) to the cap structure. Binding of the mRNA to ribosomes, the eukaryotic initiation factors eIF4E and eIF4G, and poly(A)-binding protein, results in the formation of circular structures and active translation. Step 4: termination of translation by degradation of mRNAs is catalysed by exonucleases. The cap is hydrolysed by the scavenger decapping enzymes DCP1, DCP2 and DCPS32, followed by digestion of the residual mRNA by 5′–3′ exoribonuclease 1 (XRN1). Degradation may be delayed if the mRNA is silenced and resides in cytoplasmic processing bodies. Alternatively, endonucleolytic cleavage of mRNA in the exosome may occur. The catabolism of abberant mRNA (for example, mRNA with a premature stop codon) is controlled by various other mechanisms. Step 5: the translated protein product undergoes post-translational modification, the nature of which depends on the properties of the host cell. The translated protein can then act in the cell in which it has been generated. Step 6: alternatively, the protein product is secreted and may act via autocrine, paracrine or endocrine mechanisms. Step 7: for immunotherapeutic use of mRNA, the protein product needs to be degraded into antigenic peptide epitopes. These peptide epitopes are loaded onto major histocompatibility complex (MHC) molecules, which ensure surface presentation of these antigens to immune effector cells. Cytoplasmic proteins are proteasomally degraded and routed to the endoplasmic reticulum where they are loaded on MHC class I molecules to be presented to CD8+ cytotoxic T lymphocytes. MHC class I molecules are expressed by almost all cells. Step 8: in antigen-presenting cells, to obtain cognate T cell help for a more potent and sustainable immune response, the protein product needs to be routed to MHC class II loading compartments. This can be accomplished by incorporating routing signal-encoding sequences into the mRNA. Moreover, exogenous antigens that are taken up by dendritic cells can also be processed and loaded onto MHC class I molecules by a mechanism that is known as cross-priming. Step 9: protein-derived epitopes can then be presented on the cell surface by both MHC class I and MHC class II molecules. https://www.nature.com/nrd/journal/v13/n10/full/nrd4278.html
The company uses RNA lipoplexes to selectively target the mRNA-encoding new-antigens to dendritic cells for expression and presentation to T-cells. In a paper published in Nature earlier this year, the company demonstrated the feasibility of the approach:
Lymphoid organs, in which antigen presenting cells (APCs) are in close proximity to T cells, are the ideal microenvironment for efficient priming and amplification of T-cell responses. However, the systemic delivery of vaccine antigens into dendritic cells (DCs) is hampered by various technical challenges. Here we show that DCs can be targeted precisely and effectively in vivo using intravenously administered RNA-lipoplexes (RNA-LPX) based on well-known lipid carriers by optimally adjusting net charge, without the need for functionalization of particles with molecular ligands. The LPX protects RNA from extracellular ribonucleases and mediates its efficient uptake and expression of the encoded antigen by DC populations and macrophages in various lymphoid compartments. RNA-LPX triggers interferon-α (IFNα) release by plasmacytoid DCs and macrophages. Consequently, DC maturation in situ and inflammatory immune mechanisms reminiscent of those in the early systemic phase of viral infection are activated. We show that RNA-LPX encoding viral or mutant neo-antigens or endogenous self-antigens induce strong effector and memory T-cell responses, and mediate potent IFNα-dependent rejection of progressive tumours. A phase I dose-escalation trial testing RNA-LPX that encode shared tumour antigens is ongoing. In the first three melanoma patients treated at a low-dose level, IFNα and strong antigen-specific T-cell responses were induced, supporting the identified mode of action and potency. As any polypeptide-based antigen can be encoded as RNA, RNA-LPX represent a universally applicable vaccine class for systemic DC targeting and synchronized induction of both highly potent adaptive as well as type-I-IFN-mediated innate immune mechanisms for cancer immunotherapy.
The RNA lipoplex technology is patented. We previously discussed the technology on this blog. DCs engulf naked RNA injected into lymph nodes by macropinocytosis, which is constitutively active in immature DCs. Intravenously administered RNA-LPX nanoparticles taken up by monocyte-derived human immature DCs almost completely co-localized with the macropinosome marker dextran. This allows for the cancer antigens encoded in the mRNA to be translated and epitopes expressed on MHC Class I and II molecules of the dendritic cell, stimulating antigen-specific adaptive immune responses, including CTL (cytotoxic T-cell responses), as with viral infections.
RNA-LPX nanoparticles also bind to Toll Like Receptor 3 and 7 (TLR3 and TLR7) in the endosomal compartment; this precipitates an anti-viral response from plasmacytoid dendritic cells (pDCs). IFNα is typically produced in the context of RNA virus infections by APCs (antigen presenting cells) sensing dsRNA and ssRNA via endosomal TLR3 and TLR7, respectively, and is crucial for an efficient inflammatory, antiviral environment. IFNα is required for the induction of antigen-specific (adaptive) immunity against viral infections. Therefore, RNA vaccines provide for a dual mechanism of immune modulation – adaptive and innate immune responses.
In a Nature paper published in June 2016, the company showed for the first time that precise DC targeting in lymphoid compartments can be accomplished without conjugation of RNA to ligands that bind to DC receptors, rather, by using well-known lipid carriers such as DOTMA, DOTAP, DOPE and cholesterol, without functionalization, solely by adjusting negative net charge of the nanoparticles. Negatively charged lipoplexes are preferentially targeted to the spleen following intravenous infusion, with expression of proteins encoded by mRNA in the lipoplexes.
A phase I dose-escalation trial with good manufacturing practise (GMP)-produced RNA-LPX vaccines encoding four tumour antigens (NY-ESO-1, MAGE-A3, tyrosinase and TPTE) for i.v. administration is currently recruiting patients with advanced malignant melanoma (NCT02410733, Figure 4). The first three patients were treated with a very low initial dose of RNA-LPX, followed by four weekly applications with moderately higher doses, but still below the absolute therapeutic dose levels used in mice. All vaccine applications were well-tolerated with transient flu-like symptoms. All three patients had dose-dependent early release of IFNα and IP-10 (also known as CXCL10) peaking at 6 h, resembling the kinetics observed in mice. All patients developed de novo T-cell responses against the vaccine antigens. In patient 1, who had no T cells against NY-ESO-1 at baseline, cell counts of de novo induced NY-ESO-1-specific T cells four weeks after the last immunization reached the same range as those against immune-dominant HLA-class-I-restricted peptides from cytomegalovirus, Epstein–Barr and influenza viruses. HLA-B35 NY-ESO-1 dextramer analysis of blood samples showed a rapid induction of antigen-specific CD8+ T cells within two weeks of starting treatment, increasing with subsequent vaccinations. Moreover, the pre-existing T-cell response against tyrosinase in this patient was augmented. Imaging before and after vaccination showed regression of a suspected metastatic thoracic lymph node lesion (data not shown). Patient 2, whose metastases were surgically removed before vaccination, experienced induction of CD4+T-cell responses against NY-ESO-1 and MAGE-A3 and was tumour-free at the time of this report (seven months after the start of vaccination). In patient 3, a strong NY-ESO-1-specific HLA-Cw03-directed de novo T-cell response and a weaker response against MAGE-A3 were induced. This patient previously received various treatments and had eight lung metastases at recruitment, which remained clinically and radiologically stable.
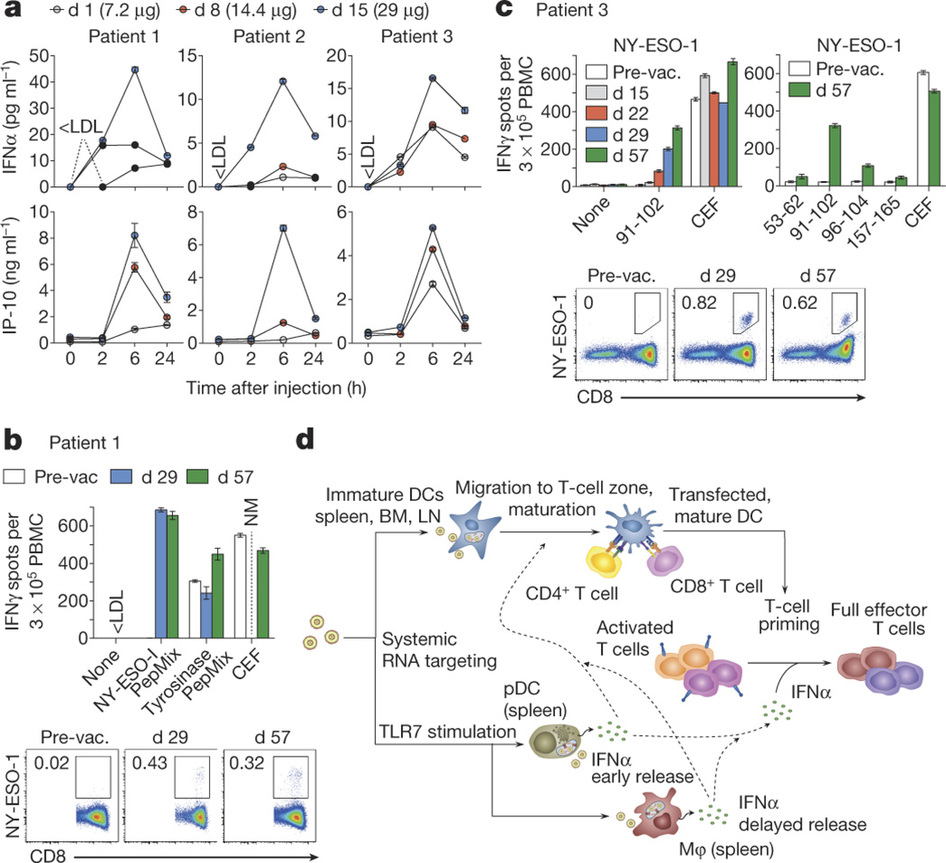
Figure 4 – Clinically administered RNA-LPX vaccines dose-dependently induce systemic INFα and de novo priming and amplification of T cells against vaccine antigens. a, Serum cytokines before (0 h) and after injection of intra-patiently escalated doses. b, c, T-cell responses against NY-ESO-1 and tyrosinase determined by restimulation with overlapping peptide mixtures or NY-ESO-1 epitopes (indicated with the amino acid position) in IFNγ ELISPOT and NY-ESO-1 specific MHC class I dextramer staining for patients 1 (b) and 3 (c). CEF, cytomegalovirus, Epstein–Barr and influenza viruses peptide pool; NM, not measured; PepMix, peptide mixture; pre-vac., pre-vaccination. d, Mechanism of action for RNA-LPX. Error bars, mean ± s.e.m. https://www.nature.com/nature/journal/v534/n7607/full/nature18300.html
TLR7-driven type-I interferon release is essential for the full anti-tumour potency of this vaccine class. Type I IFN as a key molecule in antigen-specific immunity against viral infections has further effects, for example, upregulation of MHC expression, promotion of maturation and cross-presentation in DCs, and direct inhibition of regulatory T-cell functions. The findings connect effective cancer immunotherapy with host pathogen-defence mechanisms. Mechanisms of antiviral host defense are important for survival, conserved in all vertebrates and evolutionarily optimized for high sensitivity and potency. RNA-LPX vaccines appear to mimic infectious non-self and thus mobilize concomitantly adaptive and innate antiviral mechanisms. The i.v. delivery of RNA-LPX simulates a viremic pathogen intrusion in the blood stream, and by reaching DCs in various lymphoid tissues, mobilizes the full T-cell repertoire for adaptive immune responses.
What is the rational for combining mutanome-based mRNA vaccines and checkpoint inhibitors?
Using novel antigens derived from individual patients increases the possibility that the immune response that is precipitated by vaccine strategies will result in tumor killing in individual patients. Combining this with checkpoint inhibitors will extend the duration and intensity of the immune response that is triggered. The data above included constructs that contained four known tumor antigens – NY-ESO-1, MAGE-A3, tyrosinase and TPTE – against which the immune response in patients is likely abrogated. Therefore, the collaboration with Roche will utilize mutanomes from specific patients so that novel antigens (neo-epitopes) against which a significant immune response has not yet been launched. The vaccination combined with atezolizumab is hoped to elicit a pronounced and sustained immune response to eradicate all cancer cells possessing the neo-epitopes in the mutanome, as well as other cancer cells through presentation of neo-antigens that are spilled subsequent to bystander killing.
The company is targeting melanoma, head and neck cancer, and breast cancer (Figure 5).