

Individuals that carry mutations to the CDKN2A tumor suppressor gene have 65-fold increased risk of developing melanoma and a lifetime penetrance of melanoma of 60-90%. In a new study by researchers from the Karolinska University Hospital in Sweden, individuals who had inherited CDKN2A mutations were on average 10 years younger at their melanoma diagnosis than the non-mutated familial melanoma cases.
“Despite the fact that lower age at diagnosis usually is a protective factor, these patients run more than twice the risk of dying from their melanoma,” said Hildur Helgadottir, MD, the lead researcher, in an interview. “Carriers were also seven times more likely to die of other cancers. The shorter survival time among mutation-carriers was independent of age of onset, sex, and the thickness of the skin melanoma.”
What is CDKN2A?
CDKN2A (Cyclin-Dependent Kinase Inhibitor 2A) gene encodes two gene products – INK4A/p16 and ARF/p14 (see Figure 1). This gene generates several transcript variants which differ in their first exons. At least three alternatively spliced variants encoding distinct proteins have been reported, two of which encode structurally related isoforms known to function as inhibitors of CDK4 kinase. The remaining transcript includes an alternate first exon located 20 Kb upstream of the remainder of the gene; this transcript contains an alternate open reading frame (ARF) that specifies a protein which is structurally unrelated to the products of the other variants. This ARF product functions as a stabilizer of the tumor suppressor protein p53 as it can interact with, and sequester, the E3 ubiquitin-protein ligase MDM2, a protein responsible for the degradation of p53. In spite of the structural and functional differences, the CDK inhibitor isoforms and the ARF product encoded by this gene, through the regulatory roles of CDK4 and p53 in cell cycle G1 progression, share a common functionality in cell cycle G1 control.
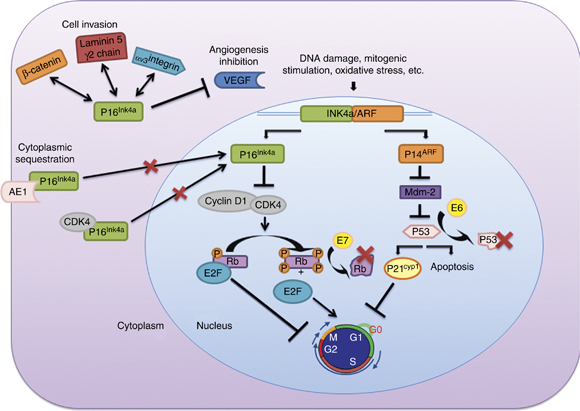
Figure 1 – Functions and interactions of the proteins coded by the INK4a/ARF locus. Different aspects of p16Ink4a molecular pathways are explained in this figure: (i) Members of the INK4 family bind and inactivate CDK4/6, blocking phosphorylation of Rb and inducing cell cycle arrest. ARF inhibits MDM2, resulting in p53 stabilization. p53 stabilization initiates cell apoptosis and/or indirectly cell cycle arrest. (ii) The molecular mechanism that explains p16Ink4a overexpression in HPV-related neoplasms is the presence of viral oncoproteins E6 and E7. Rb protein is inactivated by interaction with the high-risk HPV oncoprotein E7, and oncoprotein E6 induces degradation of the tumor suppressor p53. (iii) Cytoplasmic overexpression of p16Ink4a has been associated with its sequestration by other proteins such as CDK4 or AE1. (iv) The p16Ink4aprotein interaction with other proteins like γ2 chain of laminin 5, β-catenin or VEGF seems to be related to new functions attributable to p16Ink4a (inhibition of angiogenesis and cell invasion). http://www.nature.com/onc/journal/v30/n18/fig_tab/onc2010614f1.html
The INK4a/ARF locus encodes 2 overlapping proteins, p16 INK4a and p14 ARF, by using different first exons and common second and third exons (see Figure 2). These structurally very different proteins both act as negative regulators of the cell cycle, p16 INK4a inhibits the activation of CDK4 and CDK6 by cyclin D, hence preventing subsequent phosphorylation of pRB and thus cell cycle progression. p14 ARF regulates p53 activity by binding with MDM2, an ubiquitin ligase that otherwise targets p53 for its degradation by proteasome. High levels of p14 ARF stabilize p53 permitting it to induce p21 CIP1, a cell cycle inhibitor that blocks CDK2/cyclin E-mediated phosphorylation of pRB.
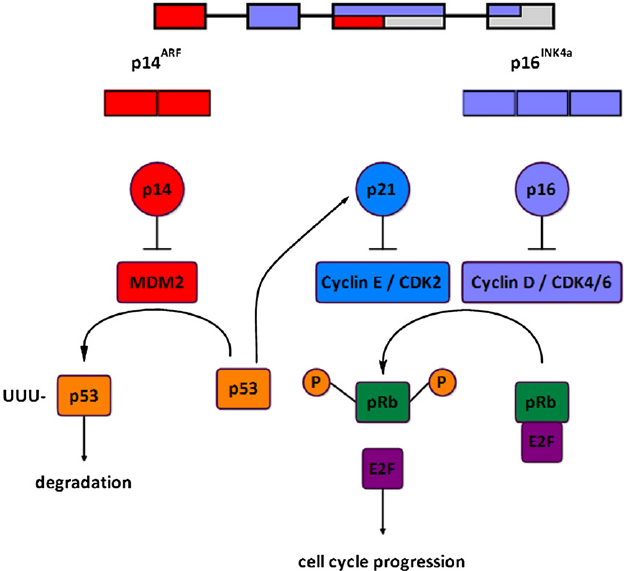
Figure 2 – The INK4a/ARF locus and cell cycle control. https://www.researchgate.net/figure/241124615_fig1_Fig-1-The-INK4aARF-locus-and-cell-cycle-control-The-INK4aARF-locus-encodes-2
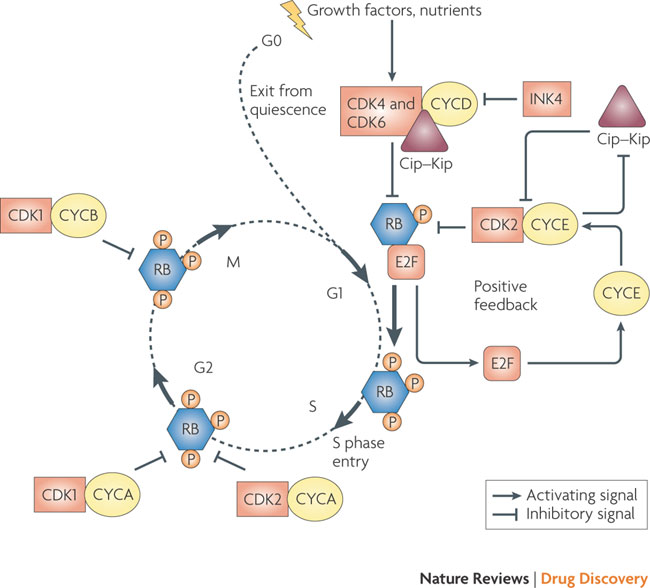
The G1 to S phase transition is a complex process, involving the concerted actions of various cyclins and cyclin-dependent kinases (CDKs) in conjunction with the RB proteins (see Figure 3). Mitogenic stimuli induce the release of the cyclin D-associated kinases, CDK4 and CDK6, from the inhibitory INK4 proteins (p16INK4A, p15INK4B, p18INK4C and p19INK4D) and initiate phosphorylation of RB1, RBL1 and RBL2. Cyclin D–CDK4 and cyclin D–CDK6 complexes also bind stoichiometrically to the potent CDK2 inhibitors of the Cip–Kip family (p27 (also known as CDKN1B and KIP1), p21 (also known as CDKN1A and CIP1) and p57 (also known as CDKN1C and KIP2)), sequestering them away from CDK2. Partially phosphorylated RB proteins release E2F transcription factors, enabling the expression of genes required for G1 to S phase transition and DNA synthesis. This includes the cyclin E gene, the protein product of which binds and allosterically regulates CDK2 activity in late G1, creating a positive feedback loop that antagonizes Cip–Kip inhibitors by signaling for their proteolysis, and reinforces RB inactivation, leading to an irreversible switch to S phase. Cyclin A- and cyclin B-dependent CDKs are activated at later phases of the cell cycle to maintain RB in a hyperphosphorylated form until the cell exits mitosis. RB family members and Cip–Kip proteins may also be involved in maintaining cells in a quiescent state (G0).
ARF, MDM2 and p53
ARF protein interferes with the activity of MDM2, leading to p53 stabilization and the triggering of a complex p53-dependent transcriptional program mediated by hundreds of target genes. MDM2 is not only a negative regulator of p53-dependent transcription and turnover, but is also a canonical p53-activated gene that has a key role in negative-feedback regulation of the p53 response (see Figure 4).
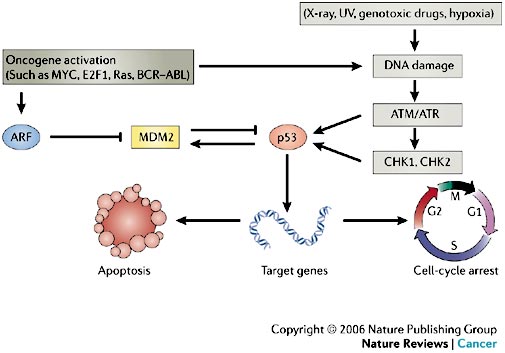
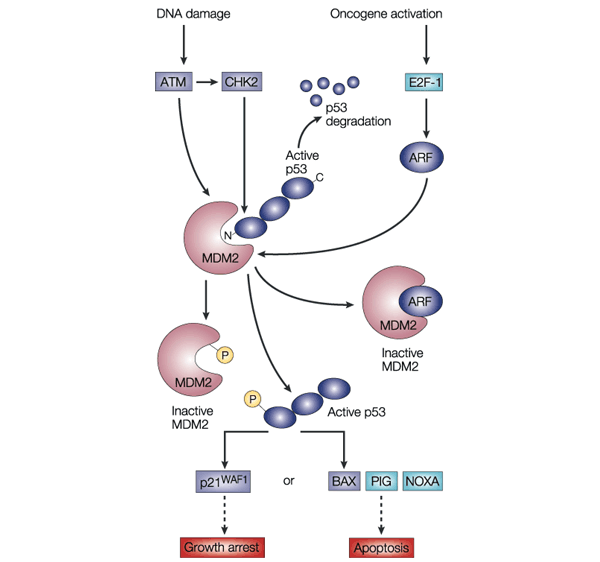
In normal cells, the transcription factor p53 is inactivated by MDM2, a ubiquitin ligase that targets p53 for degradation in the proteasome and also conceals the transactivation domain of p53 (see Figure 5). Several types of stress can activate p53, including DNA damage and oncogene activation (see Figure 5), hypoxia, depletion of the cell’s nucleotide pool or defects in DNA methylation. Each type of stress is communicated to p53 by distinct mechanisms: p53 is the master switch that integrates signals from these pathways and transforms them into a second series of signals that trigger a cellular response. This switch seems to be flipped by many post-translational modifications. For example, DNA damage triggers inactivation of MDM2 through phosphorylation of p53 and MDM2, leading to dissociation of the p53–MDM2 complex. This phosphorylation is catalysed by several kinases, including ATM and CHK2; the germ-line inactivation of these two kinases has been associated with cancer predisposition (ataxia telangiectasia and Li–Fraumeni syndrome, respectively).
What modulates melanoma penetrance in CDKN2A carriers?
The highest penetrance of melanoma in CDKN2A mutation carriers has been identified in Australia, which also has the highest incidences of sporadic melanoma worldwide. “This indicates that risk-modifying genes — associated with skin type — as well as environmental factors, such as UV-exposure, affect the impact of the CDKN2A mutation on melanoma susceptibility,” Helgadottir said.
Helgadottir recommends that members of families who have been identified as melanoma families should be offered CDKN2A mutation testing: “Families with three or more diagnoses of melanoma and/or pancreatic cancer among close relatives should be considered for CDKN2A screening. Tracing the mutation within a pedigree helps to identify individuals who benefit from screening for melanoma and other cancers.”
In particular, CDKN2A mutation carriers have a high risk of pancreatic cancers. “MRI-based screening among CDKN2A mutation carriers has been shown to result in higher rates of resectable pancreatic tumors and higher survival rates compared with sporadic pancreatic cancer cases. CDKN2A mutation carriers should also be informed about the importance of abstinence from tobacco smoke to reduce their risk of tobacco-associated cancers. Carriers who smoke should be offered smoking-cessation aids, and low-dose CT scans for screening of lung cancer should also be considered.”
I think Founndation Medicine or other laboratory (Neogenomics or Nanostring) offer it.
How do we obtain testing for CDKN2A mutation?
We live in Western Australia.