
There are two major subtypes of lung cancer: Non-Small Cell Lung Cancer (NSCLC), which accounts for 85% of all cases, and Small Cell Lung Cancer (SMLC). About 60% of NSCLC are unresectable at diagnosis, hence, the poor prognosis – ten to twelve months survival when treated with platinum-based chemotherapy. Treatment options are evaluated based on the histologic subtype and the presence of mutations to determine the the best combination of molecular therapies for treatment. Ten to twenty percent of patients with NSCLC have a mutated epidermal growth factor receptor, most commonly. a deletion in the in-frame of exon 19 (around amino acid 747 to 752) or a L858R point mutation of exon 21. On June 1, 2016, the FDA approved the first blood test (liquid biopsy) companion diagnostic to determine whether these mutations are present.

Epidermal growth factor receptor (EGFR)-mutant lung cancer, breakpoint cluster region (BCR)–ABL-driven chronic myelogenous leukaemia (CML) and KIT-mutant gastrointestinal stromal tumour (GIST) have all been treated effectively with specific tyrosine kinase inhibitors (TKIs); that is, gefitinib or erlotinib for lung cancer, imatinib for CML and imatinib for GIST. Activating drug-sensitive mutations are shown on the top of EGFR. TKI-resistant mutations are depicted on the bottom of each kinase domain schematic. The most common activating mutations in EGFR are a point mutation in exon 21, which substitutes an arginine for a leucine (L858R), and a small deletion in exon 19 that removes four amino acids (LREA). Together, these genomic changes account for ~90% of TKI-sensitive mutations that are observed in EGFR-mutant tumours. Other major drug-sensitive mutations include G719X (encoded by exon 18) and L861Q (exon 21). https://www.nature.com/nrc/journal/v10/n11/fig_tab/nrc2947_F3.html
Epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor (RTKs) that is expressed on the surface of cells and transduces signals from the cell’s exterior to the cytoplasm. Ligand binding to the N-terminal ectodomain of the cognate receptor causes the receptor to dimerize and activate each monomer’s kinase to phosphorylate partner tyrosines on the C-terminal cytoplasmic domain. Phosphotyrosines allow for emission of growth signals to target proteins within the cell. Mutations that cause an overexpression of EGFR can lead to tumor promoting properties like proliferation, anti-apoptotic characteristics, angiogenesis and migration because of downstream signaling that activates signal transduction cascades.
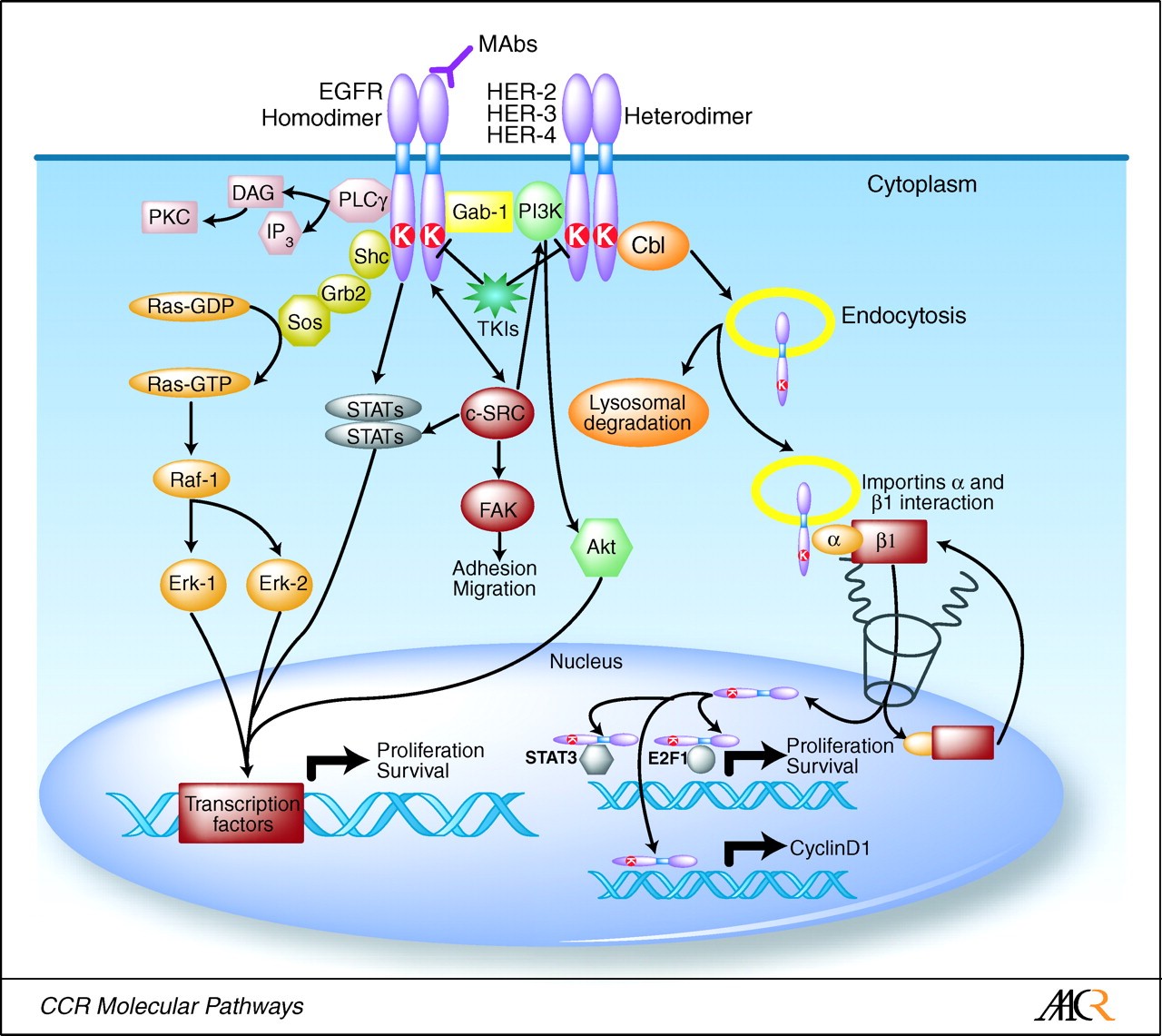
EGFR pathway, displaying downstream signal transduction cascades that are regulated by ligand binding to EGFR. https://www.pixbam.com/epidermal-growth-factor-receptor/fig.-1./461
EGFR inhibitors that have been used to target the overexpression of EGFR protein, like gefitinib, erlotinib, afatinib, lead to drug resistance; most commonly by the T790M resistance mutation. The T790M resistance occurs due to an amino acid substitution of methionine for threonine at the 790 position at exon 20. An Oncology Drug Advisory Committee voted 12 to 1 against an accelerated approval of Clovis Oncology’s rociletinib for patients that have an EGFR mutation positive NSCLC that were previously treated with an EGFR targeted therapy and tested positive for the T790M resistance mutation.
Rociletinib is Clovis’ novel, oral and irreversible mutant-selective inhibitor of EGFR. Rociletinib is a third generation EGFR inhibitor for the T790M mutation. Rociletinib blocks both the activating EGFR mutations and the T790M (which confers resistance to first and second generation TKI’s), allowing it to suppress the growth of T790M positive cells. This is important because tumors that progress following first-and second-line EGFR TKI treatment are heterogeneous, that is, comprised of clones of cells that are both T790M positive and negative (wild-type).

Intratumoral T790M heterogeneity
Panel A.
A conceptual model showing that tumors with T790M “positive” resistance to erlotinib, gefitinib or afatinib may be heterogeneous, consisting of both T790M-positive (blue) and T790 wild-type (white) populations. In some cases of rociletinib resistance, T790 wild-type cells may be the dominant driver of tumor growth and may lead to a predominantly T790 wild-type tumor (top), while in other cases the T790M-positive cells may make up the majority of the population (bottom.)
Panel B.
(I) Two representative clones out of eight isolated from cell line MGH176, derived from an afatinib-resistant pleural effusion, demonstrate heterogeneity of T790M. Brightfield images of the clones are shown on the left and nucleotide sequences on the right. Both clones are positive for the EGFR exon 19 deletion mutation. The nucleotide sequence of clone 2 reveals the presence of T790M (*), while the mutation is absent in clone 6.
(II) Summary of the single-cell clone analyses showing that overall 5 of 8 clones analyzed are T790M positive and 3 of 8 are T790 wild-type. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4497836/

Chemical structure of rociletinib
https://www.selleckchem.com/coa/co-1686-COA-S728402.html
AstraZeneca’s Tagrisso (osimertinib) received accelerated approval for patients with the T790M in November of 2015 with an overall objective response rate of 59%.


Chemical structure of osimertinib https://www.selleckchem.com/products/azd9291.html
In recommending against accelerated approval, the rociletinib panel said existing data on rociletinib did not adequately characterize its benefit-risk profile over current treatments, especially Tagrisso, and also expressed uncertainty about the proposed dose. In clinical trials, patients that were positive for EGFR-T790M mutation in metastatic NSCLC were given dosages ranging from 500 mg to 1000mg twice daily – the objective response rate across all doses was 30.2% with a mediation duration of response of 8.9 months. More than half of the patients taking any of the doses of rociletinib experienced an adverse event. Nearly half experienced a serious adverse event and 17% of the patients in the 625 mg arm died due to an adverse event. Evidence of hyperglycemia and QT prolongation–lethal heart arrhythmia–were the two leading causes of dose modification.