
TG Therapeutics is developing ublituximab, and anti-CD20 monoclonal antibody for patients with CLL (Chronic Lymphocytic Leukemia). In a phase 3 study in patients with relapsed disease, the combination of ublituximab and ibrutinib (Imbruvica) was superior to ibrutinib, alone – the overall response rates were 80% and 47%, respectively.
Other anti-CD20 monoclonal antibodies
Rituxan (rituximab) is an anti-CD20 monoclonal antibody indicated for the treatment of Non-Hodgkin’s Lymphoma (NHL), CLL, rheumatoid arthritis, and gramulomatosis. Arzerra (ofatumumab), is an anti-CD20 monoclonal antibody indicated for the treatment of CLL. Both antibodies bind to CD20 on B-cells and trigger complement-dependent cytotoxicity (CDC) and antibody-dependent, cell-mediated cytotoxicity (ADCC). Rituxan is a first generation anti-CD20 Mab – it is chimeric (mouse/human) construct. Arzerra is a second-generation anti-CD20 Mab – it is fully human.
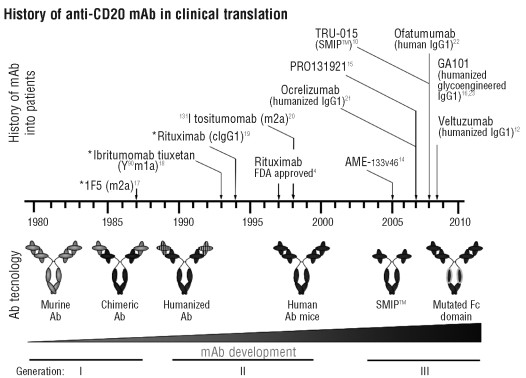
Figure 1. History of CD20 mAb in clinical translation. The time-line describes the chronological introduction over the last three decades of respective anti-CD20 mAb in human trials, as well as the corresponding progress in Ab technology from 1st through to 3rd generation reagents: generation Ab are murine or human/mouse chimeric Ab, 2nd generation Ab are either humanized or fully human and 3rd generation Ab have further modifications to the Ab structure e.g. mutation or a-fucosylation of the Fc domain for enhanced FcR binding profiles. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2805725/
Ublituximab: a third-generation anti-CD20 Mab
Ublituximab is a fully human construct characterized by a specific glycosylation pattern containing a high percentage of non-fucosylated antibody molecules at the Fc site. This specific pattern of glycosylation increases the affinity of antibodies for human FcγRIIIa (CD16), resulting in an increased antibody dependent cell-mediated cytotoxicity (ADCC) by human FcγRIIIa-expressing effector cells.
FcγRIIIa, a medium affinity receptor, capable of interaction with complexed as well as monomeric IgG, is expressed on natural killer (NK) cells, macrophages, and subsets of monocytes and γδ T cells. FcγRIIIa mediates antibody-dependent cell-mediated cytotoxicity (ADCC) by NK cells and T cells, phagocytosis by macrophages, cytokine production by NK cells and lymphocytes, and regulation of immunoglobulin production.
Antibodies that have fucose-depleted Fc portions trigger more robust ADCC:
Lec13 cells, a variant Chinese hamster ovary cell line, were used to produce human IgG1 that were deficient in fucose attached to the Asn297-linked carbohydrate but were otherwise similar to that found in IgG1 produced in normal Chinese hamster ovary cell lines and from human serum. Lack of fucose on the IgG1 had no effect on binding to human FcγRI, C1q, or the neonatal Fc receptor. Although no change in affinity was found for the His131 polymorphic form of human FcγRIIA, a slight improvement in binding was evident for FcγRIIB and the Arg131 FcγRIIA polymorphic form. In contrast, binding of the fucose-deficient IgG1 to human FcγRIIIA was improved up to 50-fold. Antibody-dependent cellular cytotoxicity assays using purified peripheral blood monocytes or natural killer cells from several donors showed enhanced cytotoxicity, especially evident at lower antibody concentrations. When combined with an IgG1 Fc protein variant that exhibited enhanced antibody-dependent cellular cytotoxicity, the lack of fucose was synergistic.
The GENUINE Phase 3 Trial
One hundred-twenty-six patients who were refractory to prior therapy and had at least one high-risk cytogenetic feature defined by the presence of 17p deletion, 11q deletion and/or p53 mutation were randomized to receive ublituximab in combination with ibrutinib or ibrutinib, alone. The chromosomal aberrations are important in the DNA damage response network of the cell. 17p (the small arm of the seventeenth chromosome) is where the p53 gene is located. p53 is the guardian of the genome; deficient p53 functioning can result in runaway cell division, which is particularly concerning when significant genomic alterations are present. 11q (the long arm of the eleventh chromosome) contains the gene ATM, which is needed to pause DNA replication until chromosomal aberrations are fixed. If repair cannot be made, p53 induces apoptosis so that the clone of cells with the damaged DNA dies off. In the absence of p53 and 11q these safeguards are not in place and cancers progress more rapidly and become more aggressive because they possess highly mutable genomes.
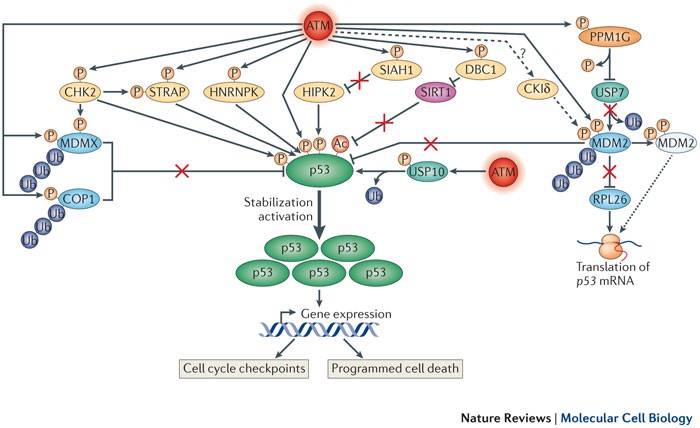
Figure 2. Within the broad DNA damage response (DDR) network, a complex but finely concerted subnetwork, governed by many ataxia- telangiectasia mutated (ATM) targets, is devoted to stimulating and increasing the amount of p53 protein, a key player in transcriptome dynamics in the face of DNA damage. For simplicity, most accessory proteins that are not under ATM control are not shown. In addition to ATM, prominent protein kinases in this network are CHK2 (checkpoint kinase 2) and HIPK2 (homeodomain-interacting protein kinase 2). Others include STRAP (Ser/Thr kinase receptor-associated protein) and HNRNPK (heterologous nuclear ribonucleoprotein K). DNA damage-induced post-translational modifications (PTMs) of p53 (which include phosphorylation, dephosphorylation and acetylation (mediated by sirtuin 1 (SIRT1)) contribute mainly to its activation and direct its transcriptional activity towards genes that are involved in cell cycle arrest and programmed cell death. The rapid stabilization and subsequent accumulation of p53 are achieved primarily by modulating the activity, stability and subcellular localization of a range of proteins. A central player in this process is MDM2, which in unprovoked cells functions as an E3 ubiquitin ligase towards p53, keeping low basal p53 expression levels in check. Following ATM- and CKIδ (casein kinase Iδ)-mediated phosphorylation, MDM2 undergoes an allosteric change that interferes with this activity. Concomitantly, ATM-mediated phosphorylation stimulates MDM2 binding to p53 mRNA, which enhances its translation (dotted arrow). This translation enhancement is also achieved by relieving MDM2-mediated polyubiquitylation of the ribosomal protein RPL26, a positive regulator of p53 translation. It is unclear whether CKIδ is stimulated by ATM (dashed arrow). Another E3 ubiquitin ligase of p53, COP1 (constitutive photomorphogenesis 1), and the p53 inhibitor MDMX undergo enhanced polyubiquitylation and proteasomal degradation following their DNA damage-induced phosphorylation by ATM, and ATM and CHK2, respectively. Two deubiquitylating enzymes (DUBS) have been found to play parts in this network to date: USP7 (ubiquitin-specific protease 7; also known as HAUSP), which alternately modulates the stability of MDM2, MDMX and p53 (not shown), and USP10, which, following its ATM-mediated phosphorylation, undergoes nuclear import and stabilization, deubiquitylates p53 and thus further contributes to its stabilization. Notably, the phosphatase WIP1 (also known as PPM1D) removes ATM-mediated phosphorylation of MDM2 and p53, thereby antagonizing this network (not shown). DBC1, deleted in breast cancer 1; SIAH1, seven in absentia homologue 1. https://www.nature.com/nrm/journal/v14/n4/fig_tab/nrm3546_F3.html