

Two companies, Celldex and Clovis, reported positive developments with their strategies to target the mutated Epidermal Growth Factor Receptor for patients with glioblastoma and lung cancer.
First, let’s take a look at the EGFR receptor to understand the biology of inhibiting ligand binding to the extracellular domain using monoclonal antibodies (Erbitux – cetuximab), and disruption of signaling from its tyrosine kinase (Iressa – gefitinib, and Tarceva – erlotinib).
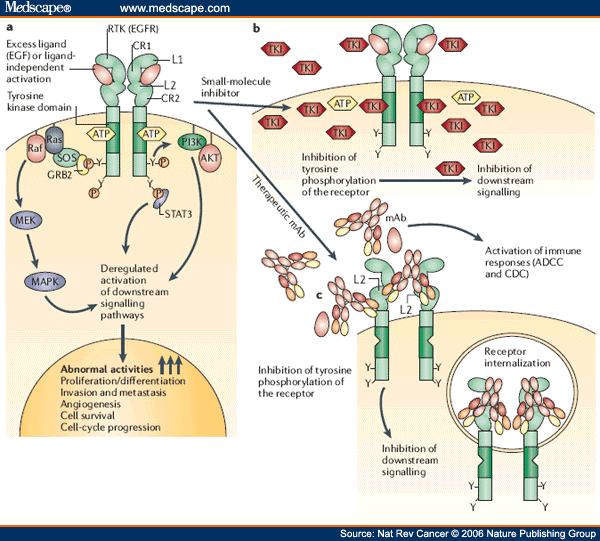
Distinct mechanisms of small-molecule inhibitors and monoclonal antibodies for targeting receptor tyrosine kinases in cancer cells. (a) Epidermal growth factor receptor (EGFR) and receptor tyrosine kinase (RTK)-dependent growth signaling in cancer cells. The extracellular region of EGFR consists of four domains, the ligand-binding domains (L1 and L2) and the cysteine-rich domains (CR1 and CR2), and the C-terminal domain of EGFR contains six tyrosine residues (Y; only two are depicted here for simplicity). Following the activation of EGFR by ligand binding or ligand-independent dimerization, the Ras–Raf–MEK–MAPK pathway is activated through the growth factor receptor-bound protein 2 (GRB2)–SOS complex. EGFR-mediated signaling also activates the phosphatidylinositol 3-kinase (PI3K)–AKT pathway, which contributes to anti-apoptotic effects of EGFR activation. Additionally, signal transducer and activator of transcription (Stat) proteins (STAT1, STAT3 and STAT5) are also activated. The coordinated effects of these EGFR downstream signaling pathways lead to the induction of cellular responses including proliferation, differentiation, cell motility, adhesion and angiogenesis. The deregulation of EGFR-mediated signalling in some cancer cells leads to aberrant proliferation, invasion, metastasis and neovascularization. (b) Small-molecule tyrosine kinase inhibitors (TKIs) such as gefitinib function as ATP analogues and inhibit EGFR signalling by competing with ATP binding within the catalytic kinase domain of RTKs. As a result, the activation of various downstream signalling pathways is blocked. Each TKI has a different selectivity for RTKs, and some are dual- or multi-selective, which might provide a therapeutic advantage. (c) By contrast, therapeutic monoclonal antibodies (mAbs) bind to the ectodomain of the RTK with high specificity (for example, cetuximab binds to the L2 domain of EGFR, and thereby inhibits its downstream signalling by triggering receptor internalization and hindering ligand–receptor interaction. Unlike small-molecule inhibitors, mAbs also activate Fcγ-receptor-dependent phagocytosis or cytolysis by immune-effector cells such as neutrophils, macrophages and natural killer cells by inducing complement-dependent cytotoxicity (CDC) or antibody-dependent cellular cytotoxicity (ADCC). MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase.
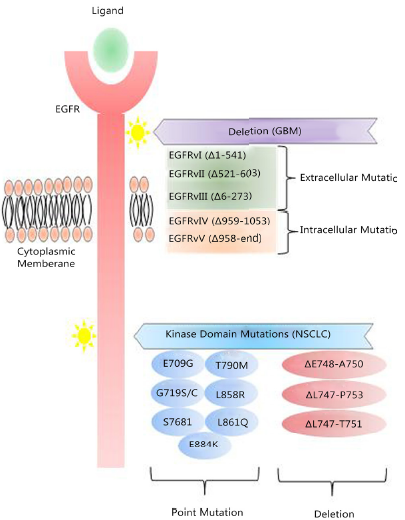
Mutations to the EGFR are responsible for relapse. A mutation of the extracellular binding domain (EGFRvIII) results in a constitutive firing of the tyrosine kinase residues that activate the Akt/PKB pathway. The T790 mutation occurs within the ATP binding cleft of the tyrosine kinase enzyme, rendering TRK inhibitors ineffective.
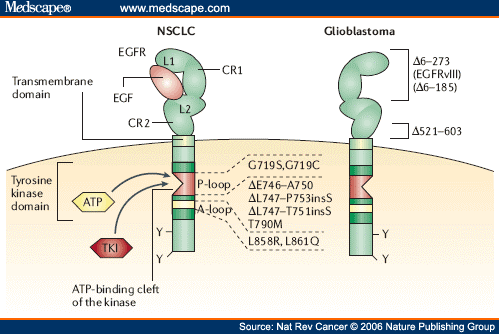
EGFR mutations correlated with clinical response to EGFR inhibitors. Two types of EGFR (epidermal growth factor receptor) mutations have been reported so far in relation to the sensitivity and resistance to gefitinib of non-small-cell lung cancer (NSCLC; left), both of which occur in the ATP-binding cleft. First, missense mutations that are detected within the nucleotide triphosphate binding domain (P-loop, exon 18; red) of the tyrosine kinase (G719S and G719C); or within the activating loop (A-loop, exon 21; yellow) (L858R and L861Q). Second, in-frame deletions with or without the insertion of a serine residue (exon 19), which are clustered in the region between codon 746–759; for example, ΔE746–A750, ΔL747–T751insS, ΔL747–P753insS. Mutations clustered within the ATP-binding cleft would be predicted to stabilize the interaction of ATP or an inhibitor molecule with this pocket, consequently leading to the more intense and sustained activation or inhibition of EGFR than that of the wild-type receptor. However, a recent report has shown that such mutations of EGFR do not affect the binding affinity of gefitinib or erlotinib to the ATP-binding pocket of the receptor, which contrasts with other activating catalytic domain mutations that have a profound effect on the interaction with imatinib mesylate, another small-molecule inhibitor. On the other hand, a resistance-related mutation, T790M, was also found within the ATP-binding cleft of the EGFR kinase domain. This mutation leads to steric hindrance to the accessibility of an inhibitor into the cleft due to the bulkiness of the methionine side chain. Unlike NSCLC, glioblastomas (right) do not frequently have mutations in the EGFR kinase domain but rather in the extracellular domain of EGFR. A recent study showed that in glioblastomas, EGFRvIII, a constitutively active genomic-deletion variant of EGFR (Δ6–273), preferentially activates the phosphatidylinositol 3-kinase (PI3K)– AKT pathway and, in tumours with intact PTEN expression, confers sensitivity to EGFR kinase inhibitors. Other EGFR mutations reported in glioblastomas include the deletion of exons 14–15, which leads to the expression of a short-form mutant partly lacking the CR2 domain (Δ521–603). However, the functional role of this mutant form remains unknown. CR1, cysteine-rich domain 1; L1, ligand-binding domain 1; TKI, tyrosine kinase inhibitor.
Tumor-type specific mutations of EGFR are well-established. In GBM, EGFR undergoes deletion processes that lead to the expression of truncated mutants, of which EGFRvIII is the major form that is associated with a poor response to conventional and EGFR-TKIs therapies. Point mutations of the EGFR kinase domain are predominant in NSCLC.
Celldex reported the following Phase 2 data on its vaccine rindopepimut (against EGFRvIII) in combination with Avastin (bevacizumab – anti-VEGF MAb) versus Avastin alone (control) in patients with EGFRvIII+ recurrent glioblastoma that had not previously received Avastin:
- Primary endpoint of PFS-6 (% alive without progression at 6 months)—rindopepimut 27% vs. control 11%; p=0.048
- Statistically significant overall survival benefit—hazard ratio of 0.47; p=0.0208; rindopepimut median 12.0 months vs. control median 8.8 months
- An increase in clinically meaningful tumor shrinkage
- Rindopepimut plus bevacizumab was well tolerated; no serious adverse events attributed to rindopepimut
- Early anti-EGFRvIII immune response correlates with survival benefit
In patients who failed prior Avastin treatment and received rindopepimut + Avastin, median survival of 5.1 months (versus 3.6 months for historical control) was observed. Additionally, early development of high anti-EGFRvIII titer was associated with longer survival and may be predictive for improved outcome in this refractory patient population.
The company is talking to the FDA about Accelerated Approval.
To better appreciate just how important and even historic a moment this is for recurrent GBM patients, consider the following quote from the National Brain Tumor Society:
There are only four FDA-approved therapies to treat brain tumors. And in the last 17 years there have been approximately 75 potential new drugs that have failed during the clinical trial process to prove they are safe and effective for patients, while only three that have won FDA approval. That is a 25:1 failure ratio. The number of drugs that have failed to meet the FDA’s safety and efficacy standard is unacceptable for the approximately 700,000 patients living with a brain tumor today, the 69,000 more who will be diagnosed within the next year, and their countless friends, families, and loved ones.
The only approved therapy for high grade gliomas that has shown any survival benefit was Temodar, approved in 2005. It extended patients’ OS with newly diagnosed GBM by 2.6 months, representing a 20% increase over surgery alone. However, even elevated doses showed no survival benefit for recurrent GBM patients. Then came Avastin, which showed 3 months improvement in progression free survival (NYSE:PFS), but did not prove to extend a patient’s overall survival. Only now has any therapy shown in a placebo controlled setting to extend patients’ overall survival in recurrent GBM–ever, in the history of this disease.
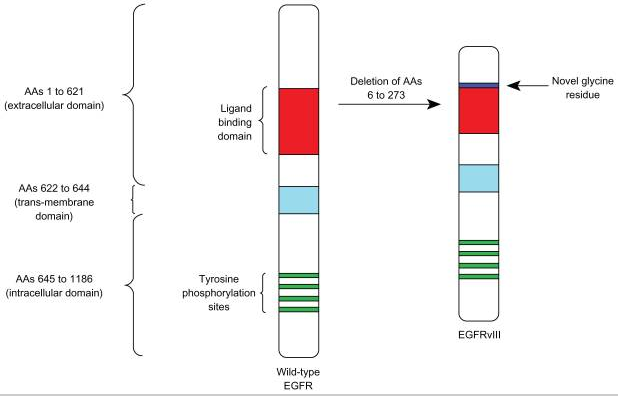
Rindopepimut is a peptide vaccine conjugated to Keyhole Limpet Hemocyanin (adjuvant), which elicits a humoral (antibody) and cell-mediated response. EGFRvIII is tumor-specific epitope expressed on approximately 20%–30% of GBMs, 78% of breast carcinomas, 73% of ovarian carcinomas, 42% of head and neck squamous cell carcinomas, and 39% of non-small-cell lung cancers. EGFRvIII expression typically occurs in the presence of wild-type EGFR overexpression, occurring in 58% to 65% of such cases. As wild-type EGFR overexpression most frequently occurs in primary (de novo) GBM and rarely in secondary GBM (those that progress from low-grade tumors), EGFRvIII expression is most common in primary tumors. Also some studies have demonstrated a male predominance for EGFRvIII expression in GBM. While EGFRvIII has various oncogenic effects, its expression in GBM has not been seen to significantly affect survival compared to those who only express wild-type EGFR. EGFRvIII contains an in-frame deletion of 801 base pairs from the extracellular domain of EGFR, resulting in the fusion of two distant portions of the molecule. This creates an antigenic junction with a novel glycine residue.20,21 This constitutively active, ligand-independent mutant enhances tumorigenicity, tumor cell migration, and resistance to chemoradiotherapy.
Clovis’ Rociletinib (CO-1686) is a novel, oral, targeted covalent (irreversible) inhibitor of the cancer-causing mutant forms of epidermal growth factor receptor (EGFR) currently being studied for the treatment of non-small cell lung cancer (NSCLC). Rociletinib was designed to selectively target both the initial activating EGFR mutations and the T790M resistance mutation, while sparing wild-type, or normal EGFR that blocks firing of the EGFR with the T970M mutation. The company is conducting studies in patients who have failed prior treatment with EGFR therapies (Erbitux, Iressa, Tarceva).
GlaxoSmithKline and Clovis will be conducting a study in patients with EGFR+ non-small cell lung cancer using rociletinib and GSK’s Mekinist (trametinib), which is a MEK inhibitor (blocking the B-raf mitogenic pathway). Another mechanism of acquired resistance in EGFR mutant NSCLC is through the activation of the mitogen-activated protein kinase (MAPK) pathway, independent of EGFR ligand binding. Trametinib is an orally active inhibitor of mitogen-activated protein kinase (MEK), which plays a key role in downstream MAPK pathway signaling, and it thereby inhibits growth factor-mediated signaling and cellular proliferation. Trametinib as a single agent has been approved by the FDA for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations, as detected by an FDA-approved test. The EGFR triggers both Ras-Raf (MAPK) and Akt/PKB pathways, as seen in the figure, below.
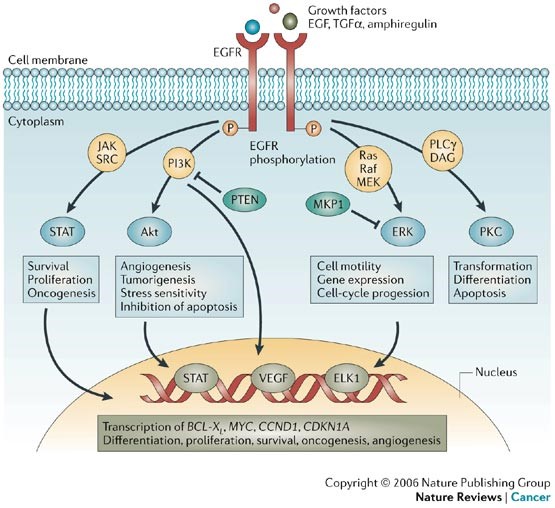
(1) Epidermal growth factor receptor (EGFR) phosphorylation can directly or indirectly activate signal transducer and activator of transcription 1 (STAT1), STAT3 and STAT5. The activated STAT proteins translocate into the nucleus and directly regulate gene expression crucial for cell survival, proliferation, transformation and oncogenesis. (2) EGFR activates phosphatidylinositol 3-kinase (PI3K) that phosphorylates phosphatidylinositol 4,5-biphosphate to form phosphatidylinositol 3,4,5-triphosphate, which then activates Akt by binding at its pleckstrin homology (PH) domain. Phosphorylated Akt has several effects, both in the cytoplasm and in the nucleus, which include the inhibition of proapoptotic factors such as BAD (BCL2 antagonist of cell death), procaspase-9 and the Forkhead (FKHR) family of transcription factors (FOXO). Akt-mediated activation of mammalian target of rapamycin (mTOR) is also important in stimulating cell proliferation, and vascular endothelial growth factor (VEGF) and hypoxia inducible factor-1alpha (HIF1alpha) are important in angiogenesis. (3) The Ras –MAPK (mitogen-activated protein kinase) pathway is activated after the activation of EGFR through two mechanisms, either through the direct recruitment of GRB2 (growth factor receptor-bound protein 2) at the SH2-domain or through the indirect recruitment of GRB2 through the SHC adaptor. Ras is activated by phosphorylation that is mediated by son of sevenless (SOS). Activated Ras binds to Raf, which in turn triggers the phosphorylation of MEK1/2 (mitogen-activated protein kinase kinase 1/2) and ERK1/2 (extracellular signal-regulated kinases 1/2). Phosphorylated ERK1/2 translocates into the nucleus and activates various transcription factors such as ELK1. Various cytoskeletal proteins are also activated by ERK1/2. PLCgamma (phospholipase c-gamma) binds through its SH2 domain to phosphorylated EGFR tyrosine kinase to become active. Once activated, (4) PLCgamma hydrolyses phosphatidylinositol 4,5-biphosphate to diacylglycerol (DAG) and inositol triphosphate (IP3). IP3 mediates calcium release from intracellular stores, affecting a host of Ca2+-dependent enzymes, whereas DAG is a cofactor for the activation of the serine/threonine kinase protein kinase-C (PKC). The activation of PKC results in cell-cycle progression, transformation, differentiation and apoptosis.