
Cells need to accumulate multiple mutations in order to induce the 10 hallmarks of cancer:
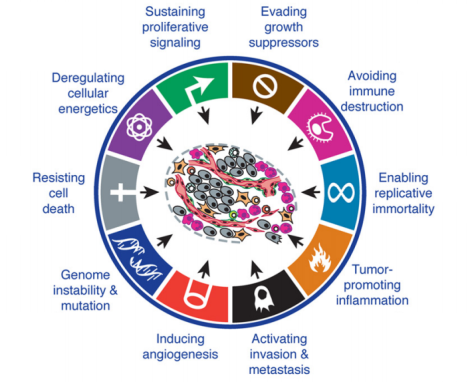
Figure 1. The ten established hallmarks of cancer. https://antisensescienceblog.wordpress.com/2014/07/17/hallmarks-of-cancer/
Accumulating the right mutations to induce the cancer phenotype takes a long time – that is why cancer is a disease of the elderly, predominantly. Moreover, cells have many DNA repair mechanisms to correct for mutations; in fact, of the 19 DNA polymerases, only 3 are for DNA replication – DNA pol α (initiation), pol d (lagging strand), and pol e (leading strand synthesis at replication fork and proofreading). Most of the other 16 DNA polymerase are for repair.
Assault with initiators, substances that induce mutation, then. are not enough to induce cancer. Agents that induce high rates of replication, that is promoters, are needed to increase the chances of acquiring the mutations and deletions. Cells appear phenotypically normal despite harboring multiple mutations. With respect to lung cancer, for example, histologically normal epithelium of lungs of current and former smokers demonstrate promoter methylation of p16 (blocker of Cyclin D – CDK4/6, which hypophosphorylates Rb as a requirement for R-point transition), mutated p53 (inducer of apoptosis), and K-ras (inducer of proliferation).
The last several mutations that induce the cancer phenotype occur more quickly than the first few, And this is greatly aided by inflammation, which induces high rates of replication of epithelial cells. Replicative stress leads to mutations and genetic events that result in oncogenic firing or oncogenes and silencing of tumor suppressor genes. The more proliferation, the more replicative stress, therefore, the greater potential for accumulating tumorigenic mutations.
Interlekin-1 induces inflammation and proliferation
Interleukin-1 (IL-1) is secreted by macrophages, B cells, and dendritic cells. Its function is to promote inflammation by activating B cells, NK cells, and T-cells. It is a pro-inflammatory cytokine that also induces proliferation via induction of NFkB.
Figure 2. IL-1 signal transduction to NF-κB activation. IL-1 binds to the IL-1R, which associates with the IL-1RAcP at the cell surface, and signals via the TIR adaptor, MyD88, through homotypic TIR-TIR interactions. Subsequently, IL-1R associated kinases (IRAKs) are recruited to the receptor/MyD88 complex. Activated IRAKs promote TNFR-associated factor 6 (TRAF6) polyubiquitination via lysine 63 linkages. The polyubiquitinated TRAF6 interacts with TAK1 in complex with TAB1 and TAB2, ultimately leading to the phosphorylation of IκBα and its dissociation from NF-κB subunits. The phosphorylated IκBα is subsequently ubiquitinated for degradation by the proteasome. The phosphorylated p65/p50 heterodimer translocates to the nucleus, where it binds to response elements in NF-kB-dependent genes and leads to the induction of pro-inflammatory gene expression. In addition, the activation of TAK1/TAB1/TAB2 leads to the phosphorylation and activation of the mitogen-activated protein kinases (MAPKs), JNK and p38. These kinases then phosphorylate and activate transcription factors, thereby inducing the expression of pro-inflammatory genes. Abbreviations: IKK, IκB kinase; IκBα, inhibitor of κB; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor κB; TAB1, TAK1 binding protein 1; TAB2, TAK1 binding protein 2; TAK1, transforming growth factorβ-activated kinase 1. https://www.sciencedirect.com/science/article/pii/S0167488914001967
CANTOS – Cardiovascular Risk Reduction Study
This study was undertaken to evaluate whether canakinumab (Ilaris), a selective, high affinity, fully human monoclonal antibody that inhibits IL-1b, could lower the risk of the development of poor cardiovascular outcomes in patients with elevated hish sensitivity c-reactive protein (hsCRP – ≥ 2mg/L), independent of LDL lipid reduction. The study was quite positive, thereby establishing the benefit of reducing inflammation in patients with cardiovascular disease.
At 48 months, the median reduction from baseline in the high-sensitivity C-reactive protein level was 26 percentage points greater in the group that received the 50-mg dose of canakinumab, 37 percentage points greater in the 150-mg group, and 41 percentage points greater in the 300-mg group than in the placebo group. Canakinumab did not reduce lipid levels from baseline. At a median follow-up of 3.7 years, the incidence rate for the primary end point was 4.50 events per 100 person-years in the placebo group, 4.11 events per 100 person-years in the 50-mg group, 3.86 events per 100 person-years in the 150-mg group, and 3.90 events per 100 person-years in the 300-mg group. The 150-mg dose, but not the other doses, met the prespecified multiplicity-adjusted threshold for statistical significance for the primary end point and the secondary end point that additionally included hospitalization for unstable angina that led to urgent revascularization (hazard ratio vs. placebo, 0.83; 95% CI, 0.73 to 0.95; P=0.005).
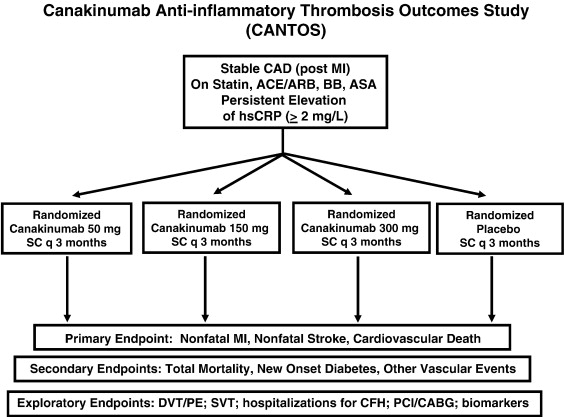
Figure 3. CANTOS study design. https://www.sciencedirect.com/science/article/pii/S0002870311004820
What does this have to do with lung cancer?
Another major finding of the CANTOS study was a substantial reduction in the rate of development of lung cancer – a relative risk reduction of 67% finding for lung cancer (HR 0.33 [95% CI: 0.18-0.59]) and 77% for lung cancer mortality (HR 0.23 [95% CI: 0.10-0.54]) observed among patients receiving the 300mg dose.
With more than 10,000 patients enrolled in the study over the last six years, CANTOS was one of the largest and longest-running clinical trials in Novartis’ history. Trial participants with a prior history of atherosclerosis, a hsCRP level of >=2mg/L, and who were free of previously-diagnosed cancer, received either placebo or one of three doses of ACZ885 (50mg, 150mg, and 300mg subcutaneously every 3 months). All participants received current standard of care therapies, with 91% of participants taking lipid-lowering statins. During a median follow up of 3.7 years, as compared to placebo, ACZ885 resulted in dose dependent reduction in hsCRP of 26 to 41% and a dose-dependent reduction in IL-6 of 25 to 43% (p=<0.0001). For all cancer related mortality (n=196 across treatment), ACZ885 resulted in a significant reduction compared to placebo at the 300mg dose (HR 0.49: [95% CI: 0.31-0.75] p=0.0009). Incident lung cancer (n=129 across treatment) was reduced at the 300mg dose versus placebo (HR 0.33 [95% CI: 0.18-0.59]; p=<0.0001) and the 150mg dose versus placebo (HR 0.61 [95% CI: 0.39-0.97]; p=0.034). Lung cancer mortality was significantly less common at the 300mg dose versus placebo (HR 0.23 [95% CI: 0.10-0.54] p=0.0002).

Figure 4. Lung cancer data – CANTOS. https://www.valuentum.com/articles/Canakinumab_Posts_Some_Impressive_Data_For_Novartis
- During the median 3·7 year follow-up period, total cancer mortality was lower in the combined canakinumab groups than in the placebo group (p=0·0158). For this endpoint (n=196), compared with placebo, HRs were 0·86 (95% CI 0·59–1·24; p=0·42) for the 50 mg group, 0·78 (95% CI 0·54–1·13; p=0·19) for the 150 mg group, and 0·49 (95% CI 0·31–0·75; p=0·0009) for the 300 mg group. The incidence rate of cancer mortality per 100 person-years was 0·64 in the placebo group, 0·55 in the 50 mg group, 0·50 in the 150 mg, and 0·31 in the 300 mg group (p=0·0007 for trend across active dose groups compared with placebo).
- Lung cancer accounted for 26% of all cancers and 47% of all cancer deaths in the placebo group, but only 16% of all cancers and 34% of cancer deaths in the canakinumab groups. For incident lung cancer (n=129), compared with placebo, HRs were 0·74 (95% CI 0·47–1·17; p=0·20) for the 50 mg group, 0·61 (95% CI 0·39–0·97; p=0·034) in the 150 mg group, and 0·33 (95% CI 0·18–0·59; p<0·0001) for the 300 mg group (figure 2B, table 2). The incidence rate of lung cancer per 100 person-years was 0·49 in the placebo group, 0·35 in the 50 mg group, 0·30 in the 150 mg group, and 0·16 in the 300 mg group (p<0·0001 for trend across active dose groups compared with placebo).
- Lung cancer mortality (n=77) was significantly less common in the canakinumab 300 mg group than in the placebo group (HR 0·23 [95% CI 0·10–0·54]; p=0·0002; figure 2C, table 2). The incidence rate of lung cancer mortality per 100 person-years was 0·30 in the placebo group, 0·20 in the 50 mg group, 0·19 in the 150 mg group, and 0·07 in the 300 mg group (p=0·0002 for trend across active dose groups compared with placebo; figure 2C, table 2).
Implications for inflammation and cancer
Canakinumab (quarterly subcutaneous injections for a median 3.7 years) successfully reduced inflammation in the coronary blood vessels and epithelium of the lung. With respect to lung cancer, patients most likely had several mutations in the epithelium of their lungs prior to entering the study (if not undetected frank cancer) , and, because all patients had elevated hsCRP, their lung epithelia were proliferating at an accelerated pace, predisposing them to accumulate additional mutations quickly:
- Twenty-four percent of the study participants were current smokers and 47% were past smokers;
- Compared with participants who were not diagnosed with cancer, those who developed incident lung cancers were older (p<0·0001) and more likely to be current smokers (p<0·0001). Patients who developed lung cancer were on average 65 years old at study entry, and more than 90% were current or former smokers.
- Median hsCRP (6·0 mg/L vs 4·2 mg/L; p<0·0001) and interleukin 6 concentrations (3·2 ng/L vs 2·6 ng/L; p<0·0001) were significantly higher at baseline in people who were diagnosed with lung cancer during follow-up than in those who remained free of any cancer diagnosis;
- Stratification by smoking suggested that the effect of canakinumab on lung cancer was slightly stronger in current than in past smokers (HR 0·50 [p=0·005]. This effect was more prominent in the 300 mg group (HR 0·25 [p=0·002] vs HR 0·44 [p=0·025].
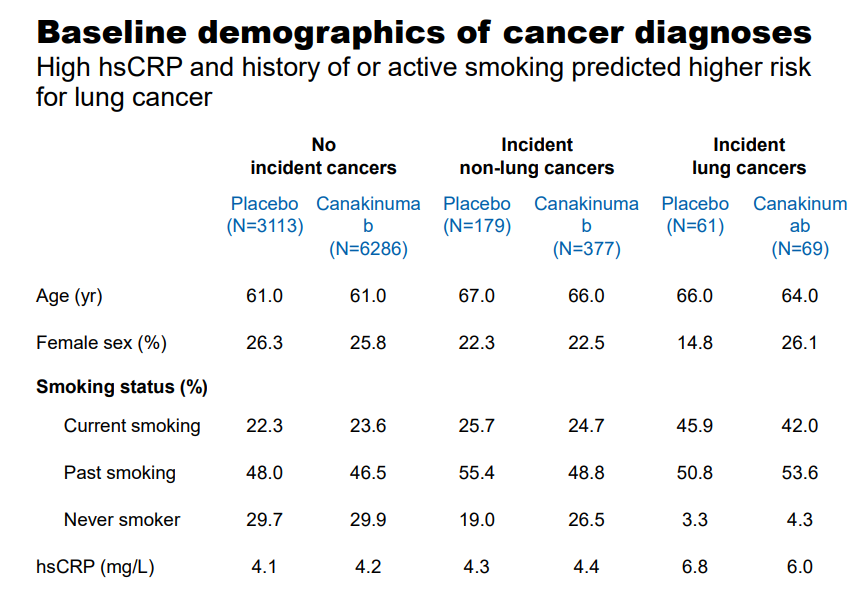
Figure 5. Baseline demographics of lung cancer diagnoses. https://www.valuentum.com/articles/Canakinumab_Posts_Some_Impressive_Data_For_Novartis
What, exactly, is canakinumab doing? There are several possibilities:
- depleting IL-1b in the microenvironment thereby diminishing the ability of cancers that were already present to grow and induce angiogenesis;
- reducing the levels of IL-1 in the tumor microenvironment essential to promoting invasiveness of cancers that were already present (though not detected), and/or
- blocking IL-1-induced inflammation, thereby, reducing the rate of proliferation of lung epithelium which would prevent the accumulation of the remaining mutations required to induce the invasive/metastatic cancer phenotype.
The CANTOS trial is truly groundbreaking. Strategies to reduce inflammation, particularly in patients with elevated hsCRP who at high risk of developing cancer should be further evaluated.