
P53 is a tumor suppressor gene that pauses cell division to allow for repair of gene damage, and triggers apoptosis if the damage is not reparable. Loss of p53 is a critical step in the evolution of cancer. Most frequently, p53 is mutated at its DNA binding domain; since p53 is a transcription factor, a diminished ability to bind to DNA significantly disrupts its functioning.
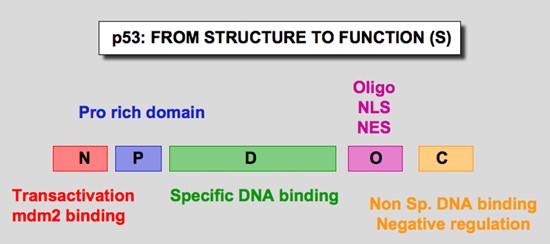
Figure 1. p53 domains. Human p53 protein (Hp53) can be divided into five domains, each corresponding to specific functions (Figure 6) . I) The amino-terminus part 1-39 contains the acidic transactivation domain and the mdm2 protein binding site. II) Region 40-100 contains series repeated proline residues that are conserved in the majority of p53. III) The central region (101-306) contains the DNA binding domain. It is the target of 90% of p53 mutations found in human cancers. IV) The oligomerization domain (307-355) consists of a beta-strand, followed by an alpha-helix necessary for dimerization, as p53 is composed of a dimer of two dimers. A nuclear export signal is localized in this oligomerization domain. V) The carboxy-terminus of p53 (356-393) contains 3 nuclear localization signals and a non-specific DNA binding domain that binds to damaged DNA. This region is also involved in downregulation of DNA binding of the central domain. https://p53.free.fr/our_work/xenopus.html
MDM2 and MDMX are regulators of p53 function – MDM2 is an ubiquitin ligase that tags p53 for destruction in the proteasome; MDMX inhibits the transcriptional activity of p53.
MDMX is commonly overexpressed in tumors, and its upregulation has been shown to promote tumor formation (Danovi et al., 2004). Unlike MDM2, however, MDMX expression is not induced by DNA damage (Shvarts et al., 1996). MDMX binds to both MDM2 (Sharp et al., 1999) and p53 (Shvarts et al., 1996). MDMX binding to MDM2 inhibits MDM2 autoubiquitination (Okamoto et al., 2009). Furthermore, MDM2 ubiquitinates MDMX (De Graaf et al., 2003). The interaction of MDMX and p53 has been shown to inhibit p53 activity (Marine et al., 2007). Figure 2 schematically depicts the relationships between p53, MDM2, and MDMX. MDMX possesses a cryptic nuclear localization signal (LeBron et al., 2006), so it can only reach the nucleus while bound to other proteins. MDMX is normally located primarily in the cytoplasm (Gu et al., 2002).
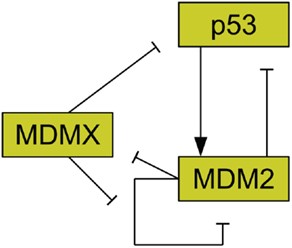
Figure 2. Relationships between MDMX, MDM2, and p53. MDM2 inhibits p53 and is promoted by it. MDM2 inhibits itself and this effect is reduced by MDMX. MDMX inhibits p53 directly, and is itself inhibited by MDM2. https://journal.frontiersin.org/article/10.3389/fonc.2013.00064/full
Disrupting the binding of MDM2 and MDMX to p53 are an excellent strategy to restore p53 functioning. However, this approach works on wild-type p53. Most p53 mutants are no longer subject to MDM2 ubiquitylation and are stable.
ALRN-6924 inhibits both MDM2 and MDMX
ALRN-6924 is a stapled peptide high-affinity decoy that attracts and binds MDMX and MDM2, thereby causing the release of the bound p53. A phase 1 trial in 71 patients with 24 different cancers (all comers) was completed in January 2017. In addition, ALRN is being developed for the treatment of Peripheral T-cell Lymphoma (PTCL), Acute Myeloid Leukemia (AML), and Myelodysplastic Syndrome (MDS). The drug has been granted Orphan Drug stsus for AML.
In the phase 1 trial, 41 patients received a >3.2 mg/kg dose of ALRN-6924 per treatment cycle and who did not have a mutation of the p53 gene:
Of the 41 patients, there were two complete responses (CR), two partial responses (PR), and 20 with stable disease (SD). The disease control rate was 59% (24/41). One CR patient, both PR patients and two SD patients have received ALRN-6924 for well over one year, the CRs and PRs are ongoing as of May 1, 2017 and patients continue to receive treatment with ALRN-6924.
Aileron is designing drugs that disrupt protein-protein interactions, traditionally considered “undruggable targets.” Its “stapled peptide” technology involves “stapling” amino acid sequences with hydrocarbon bonds that maintain their natural alpha-helical shape, the most common protein structure at the interface of protein-protein interactions .Additional targets include other transcription factors and signaling proteins such as Ras, Myc, ß-Catenin, the Bcl family of proteins and HIF-1a.