
The last twenty years has been an unprecedented time in biology – in sequencing the genome and studying the functions of proteins, as well as in unraveling signal transduction pathways, the fundamental biology of normal and diseased cells has been elucidated to a great extent. Although many druggable targets have been identified, it has largely been impossible to target protein-protein interactions (PPI) in drug development. In fact, only ONE drug that targets a PPI has been approved.
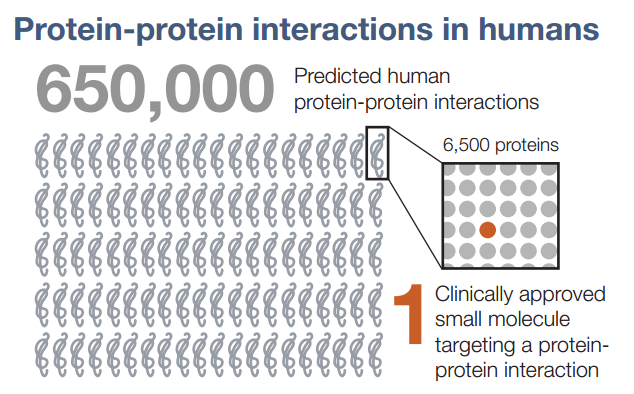
Figure 1. Protein-protein interactions – difficulty in developing drugs. https://www.cell.com/cell/pdf/S0092-8674(16)30727-9.pdf
What are protein-protein interactions?
PPI involve transient interaction between proteins that are driven by electrostatic and hydrophobic interactions, as well hydrogen bonds and Van der Waals forces. Unlike enzyme-substrate interaction that are druggable, PPI do not have well-defined and well-circumscribed binding pockets, rather, the two proteins “associate” with each other over large areas and in a less definable manner. The associations among transcription factors and their co-activators are prime examples of PPIs.
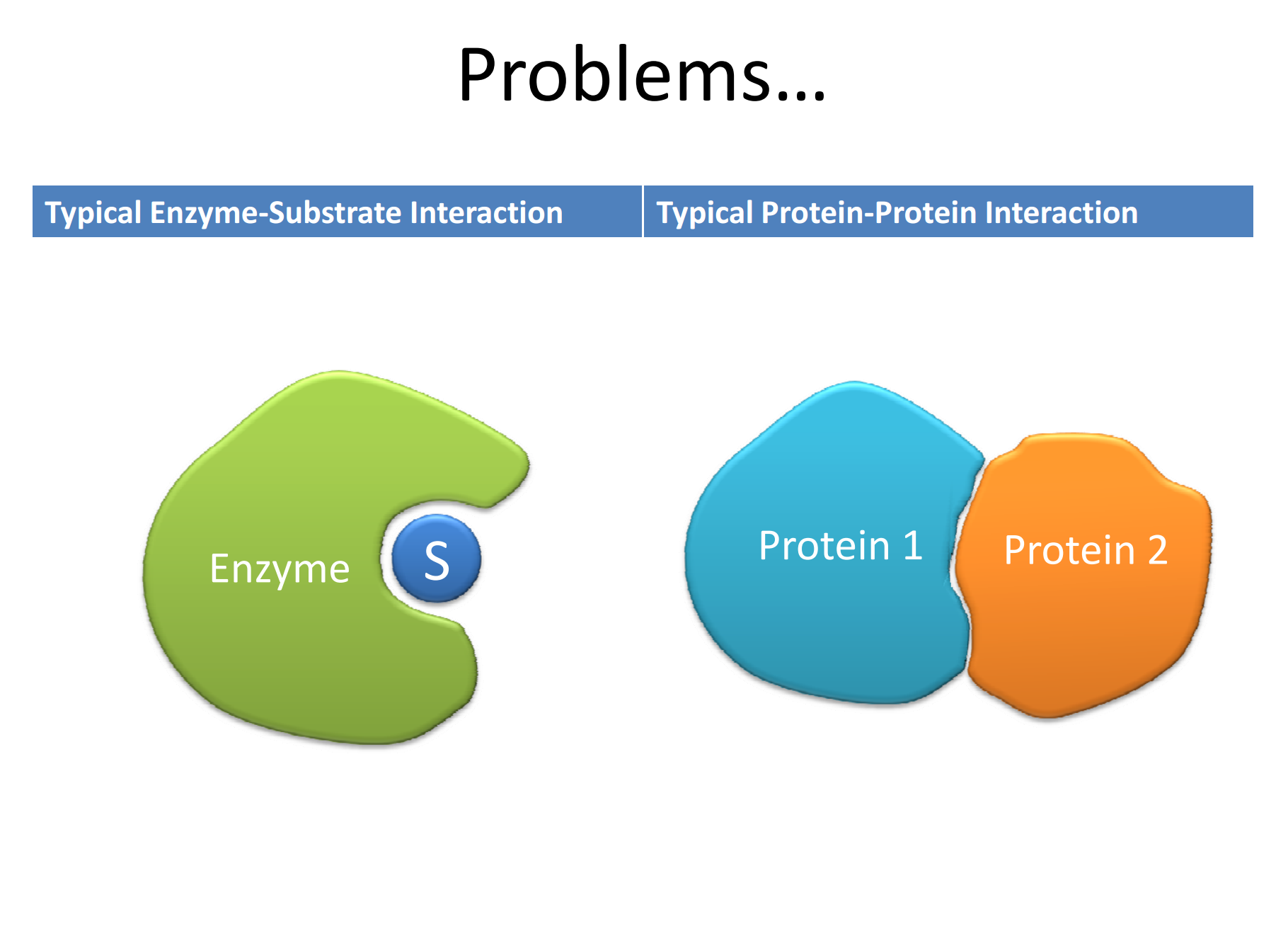
Figure 2. Enzyme-substrate versus protein-protein interactions https://workspace.imperial.ac.uk/bhfcre/Public/Teaching/2.4%20leatherbarrow.pdf
Why are protein-protein interactions difficult to target?
Because PPIs have no immediate and direct output by which to measure their occurrence (e.g., enzymatic activity), studying PPIs in vitro has proven extremely difficult.
Protein-Protein Interactions (PPIs) are the basis of many important cellular processes and being able to disrupt or stabilize PPIs offers innovative routes for new therapeutics. However, PPIs are difficult to screen against due to their lack of easily assayable output (no enzymatic activity to measure). Indeed, some pharmaceutical industry companies remain apprehensive of full-fledged drug discovery campaigns in this direction.
If assays cannot be developed, then screening libraries of compounds in an efficient and predictable manner is not possible. With PPIs, proteins interact with each other over large portions of their respective molecules. This represents a two-fold problem – (1) small molecule approaches, therefore, have not borne much fruit because they cannot span the full length of the PPI area, and (2) PPIs targets of interest inside the cell are not accessible by large molecules administered exogenously. Moreover, the interactions are not robust – they are easily disrupted by subtle changes in the vicinity of the PPI. So, quantitating drug effects in the evaluation and selection of potential drugs to modulate the interactions is extremely difficult.
Success to date
There has been just ONE approved drug that targets PPI – venetoclax (Venclexta), which inhibits the interaction between Bcl-2, a BH3-only protein, and the pro-apoptotic proteins, BAK and BAX. The drug was approved in 2016. There are no binding pockets for Bcl-2 on BAX/BAK, rather, the interaction is characterized by transient association.
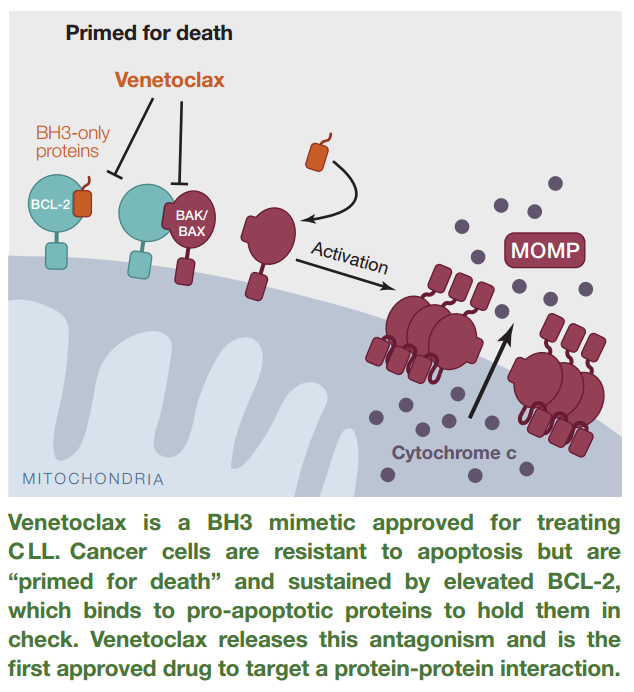
Figure 3. Venetoclax disrupts the interaction of Bcl-2 and BAK/BAX, thereby, allowing apoptosis of chronic lymphocytic leukemia cells harboring the 17p deletion. https://www.cell.com/cell/pdf/S0092-8674(16)30727-9.pdf

Figure 4. Venetoclax mechanism of action. https://www.empr.com/new-drug-product-venclexta/slideshow/3217/
Inthera Bioscience’s Approach to Protein-Protein Interactions
Inthera recently raised $11MM (including participation from Merck Ventures and Novo Seeds) to propel its development efforts. “Inthera Bioscience employs a proprietary technology platform to rationally design small molecule inhibitors of intracellular protein-protein interactions (PPIs).”
Their approach involves first identifying key binding characteristics of one of the key helical peptide interfaces in the PPI and mimicking it on an oligo-oxopiperazine backbone, which permits rational drug discovery of small molecules. Basically, a small molecule model (called an oxopiperazine helix mimetic, or Ohm) of the protein is made – this permits evaluation of drug candidates in a more reliable and quantitative manner. The Ohm is about half the size of the helical peptide that it models.
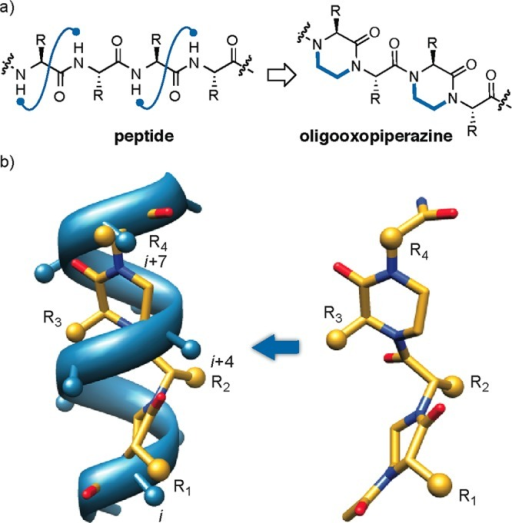
Figure 5. (a)Design of oxopiperazine helix mimetics. (b) Overlay of an 8-mercanonical α-helix and an oxopiperazine dimer (left). Predictedlow-energy structure of an oxopiperazine dimer (right). Side chaingroups are depicted as spheres. https://openi.nlm.nih.gov/detailedresult.php?img=PMC4353027_ja-2014-02310r_0002&req=4
Their first program involves reactivating the p53 tumor suppressor pathway in HPV cancers. The E6 and E7 proteins inhibit p300 from post-translational acetylation of p53. Inthera is designing molecules that prevent E6 and E7 from interacting with p300.
The CD1 domain comprises the first 20 amino acids of E7 and is critical for E7’s ability to induce S-phase progression and cellular transformation. Among the binding partners that interact with this domain of E7 are UBR4/p600 and p300/CBP-associated factor (P/CAF)..
E6 can also control p53-dependent gene regulation, via its interactions with p300/CBP co-activators. Additionally, it was demonstrated that E6-interacting regions of p300 are necessary for E6 to inhibit p53-dependent chromatin transcription, and that the E6- mediated repression of p53-dependent activation correlates with inhibition of acetylation on p53 and nucleosomal core histones, altering p53 and p300 recruitment to chromatin.
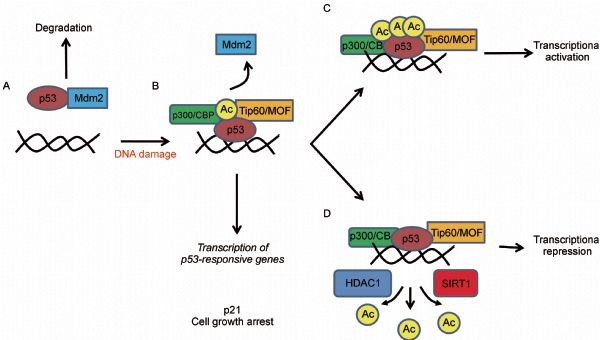
Figure 6. The role of p53 acetylation in gene regulation. (A) Unacetylated p53 is capable of activating genes that are involved in the negative regulation of p53, such as Mdm2, as a mechanism to keep p53 protein levels low during times of normal homeostasis. (B) Upon DNA damage, acetylation of p53 allows for the disruption of the Mdm2-p53 interaction and the recruitment of specific HATs to the promoters of genes involved in DNA repair and cell cycle control. (C) Full acetylation of p53 at all key acetylation sites promotes the activation of proapoptotic genes. (D) SIRT1 and HDAC1 are deacetylases that can reverse p53 acetylation and lead to transcriptional repression. https://journal.hep.com.cn/pac/EN/10.1007/s13238-011-1063-9