

Last week, Merck acquired OncoEthix at a price of $375 MM for OTX015, a synthetic small molecule which targets the BET bromodomain proteins 2, 3, and 4 (BRD2/3/4).
BRDs 2, 3, and 4 are considered potential cancer targets because of their pivotal role in regulating the transcription of growth-promoting genes and cell cycle regulators. Of particular interest, inhibition of BRD2/3/4 down-regulates MYC transcription, followed by genome-wide down-regulation of Myc-dependent target genes.
MYC is a very powerful oncogenic transcription factor, which is acts by the following mechanisms:
- Blocking inhibitors of proliferation, including p15 (INK4B), p16 (INK4A), p21 (Cip1), and p27 (Kip1);
- Activating inducers of proliferation, including Cyclin D2, E2F(1-3), Cks1, and Cul1;
- Inducing anti-differentiation bHLH transcription factors via induction of Id2;
- Inducing telomerase.
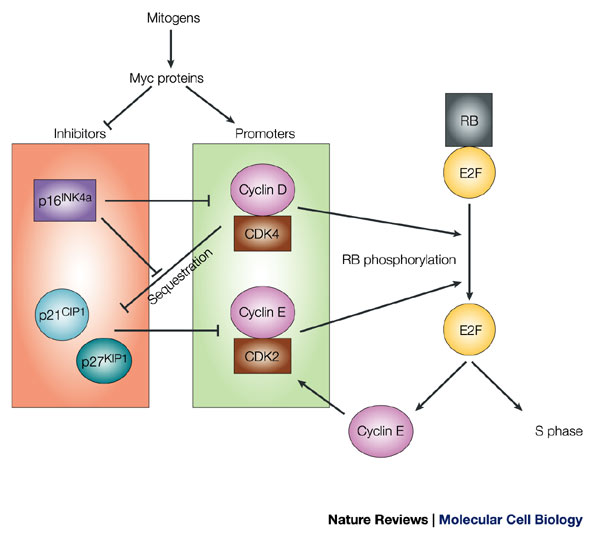
Mitogenic signals that are mediated through Myc-family proteins promote G1–S progression by several mechanisms. Myc induces the expression of cyclin D and cyclin-dependent kinase (CDK)4, which together form an active kinase complex. Low-level phosphorylation of retinoblastoma (RB) family proteins by cyclin-D–CDK4 (or cyclin-D–CDK6, not shown) disrupts interactions between RB and E2F proteins, which relieves RB-mediated repression of E2F-target genes. Early E2F-dependent transcription of cyclin E (which might also be a Myc target) leads to the formation of active cyclin-E–CDK2 complexes. Cyclin-E–CDK2 functions in a positive-feedback loop to promote RB hyperphosphorylation. This releases E2F, which results in the full activation of E2F target genes that promote entry into S-phase. The activities of cyclin–CDK complexes are opposed by CDK inhibitors, which might be targets for Myc-mediated repression. p21CIP1 and p27KIP1 inactivate cyclin-E–CDK2 complexes. p21CIP1 and p27KIP1 are also sequestered by active cyclin-D–CDK complexes, which frees cyclin-E–CDK2 from their inhibitory effects. p16INK4a binds to, and disrupts, cyclin-D–CDK complexes, which blocks G1–S progression both by preventing the cyclin-D–CDK4-mediated RB protein phosphorylation and by releasing sequestered p21CIP1 and p27KIP1. http://www.nature.com/nrm/journal/v5/n10/fig_tab/nrm1491_F1.html
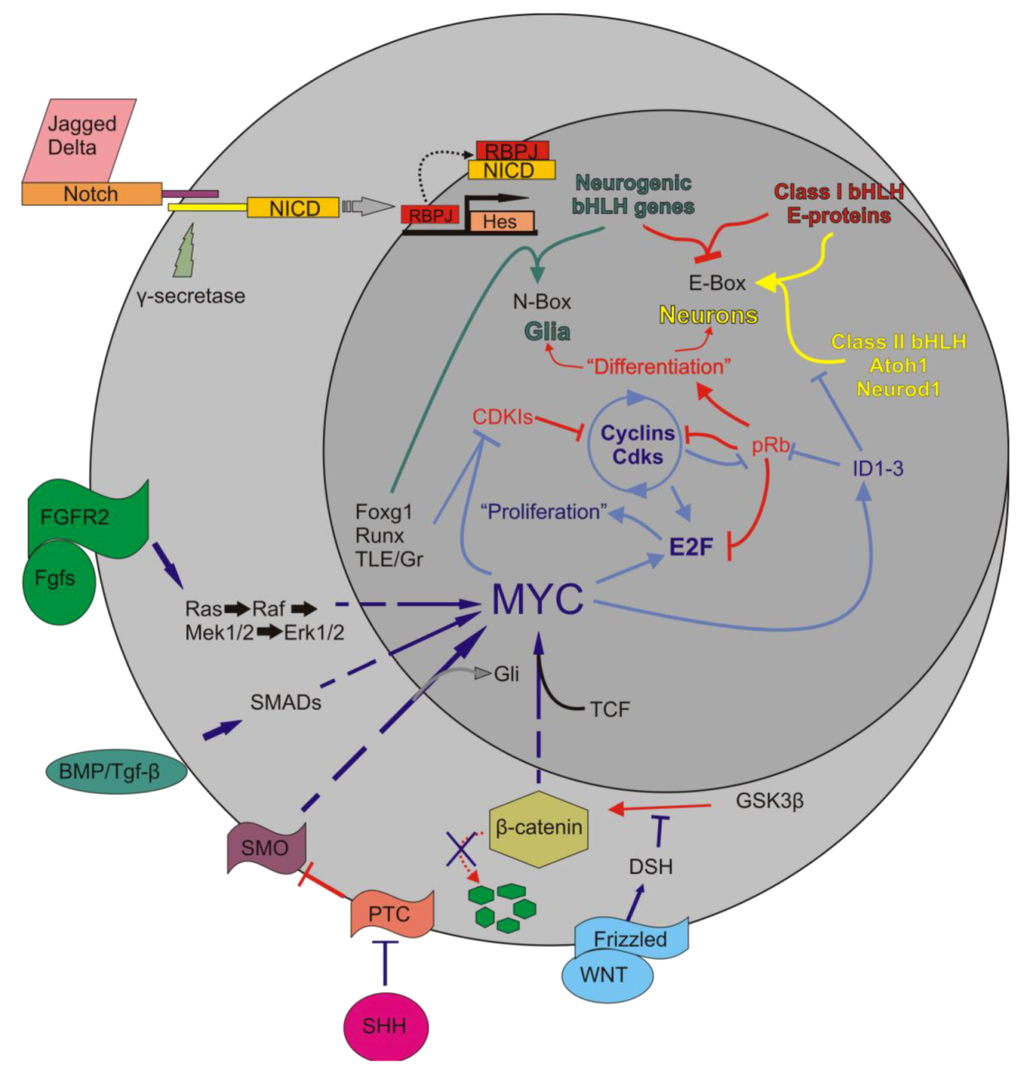
The interactions of Myc in the body are complex. Upon activation of Notch by Jagged or Delta, the Notch IntraCellular Domain (NICD) is cleaved by γ-secretase. NICD translocates to the nucleus to form a complex with RBPJ to positively regulate the transcription of the Hes family of bHLH TFs [63]. Myc is regulated by a number of pathways, including the Wnt/β-catenin pathway, Tgf-β or Bmp/Smad, Fgf/Erk1/2, and the Shh/Smo pathways. Once activated, Myc interacts with partner proteins to regulate a number of downstream effectors, including the cyclins, E2Fs, IDs, CDKIs, and various microRNAs. This regulation leads to the inhibition of class II bHLH TFs from binding to the E-proteins which then allows the Hes family to occupy the E-proteins. It also leads to the removal of pRb from E2F and allows E2F to bind S-phase promoting genes. The larger circle represents the cell as a whole while the smaller circle is the nucleus. Arrows indicate upregulation while blunted lines represent inhibition. Red indicates pathways favoring differentiation, blue indicates pathways favoring proliferation – http://www.mdpi.com/2073-4409/1/4/667/htm
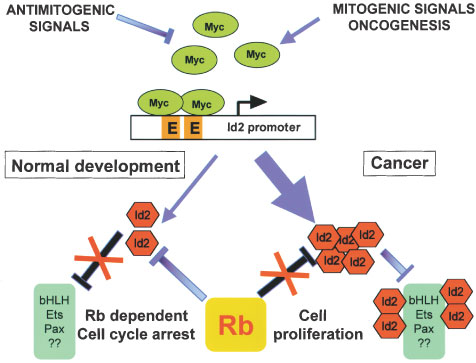
The Myc-Id2-Rb pathway in development and cancer. Myc proteins are intracellular sensors of mitogenic and antimitogenic signals. When constitutively activated by genetic alterations, Myc are transformed in powerful oncoproteins. Myc bind two E-boxes in the Id2 promoter and activate transcription. During normal development of the nervous system, activity of the N-Myc-Id2 pathway is restrained by Rb. Rb binds Id2 and might prevent Id2-mediated inhibition of bHLH, Ets, Pax and maybe other transcription factors. In neuroblastoma, N-myc amplification and overexpression induces accumulation of Id2 to levels exceeding active Rb. Id2 would be free to inhibit transcription of the natural targets that control cell proliferation. E, E-box (http://www.nature.com/onc/journal/v20/n58/fig_tab/1205093f1.html#close)
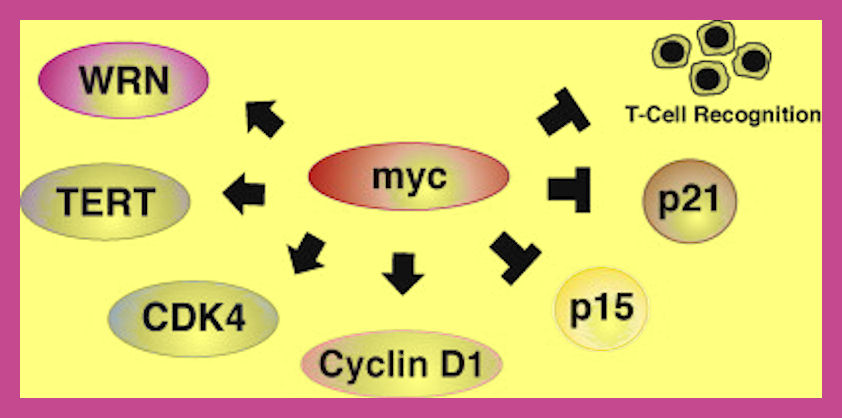
Myc induces genes conferring cell-cycle progression (e.g. CycD1 and CDK4), and represses cell-cycle inhibitors (e.g. p15INK4b, p21CIP1). Moreover, Myc drives the expression of target genes (e.g. WRN, TERT), whose inactivation unveils senescence under the condition of active Myc. In addition, Myc possesses immunosuppressive potential by impairing antigen processing and presentation, thereby preventing T-cells from recognizing tumor cells, and, thus, from releasing pro-senescent secretable factors in closest proximity to the Myc-driven cells. http://scientificearthconscientious6.wordpress.com/2012/12/12/previously-unknown-mechanism-identified-in-oncogene-induced-senescence-reported-in-the-american-journal-of-pathology/
Previously, pharmacologic inhibition of MYC—a transcription factor that is a master regulator of diverse cellular functions with a role in a range of human cancers—was not considered possible. OTX015 is the first BRD2/3/4 inhibitor to enter clinical trials.
In experimental models of Myc-dependent hematologic malignancies, including acute myeloid leukemia, Burkitt’s lymphoma, and multiple myeloma, inhibition of BRD2/3/4 produced a potent antiproliferative effect associated with cell cycle arrest and cellular senescence.
OTX015 is in Phase 1 clinical trials in patients with hematologic malignancies, advanced solid malignancies, and glioblastoma.