
Roche /Genentech announced that it acquired a company called Seragon for its new class of compound – SERD (selective Estrogen Receptor Degraders). Blocking estrogen production (aromatse inhibitors) and the binding to the estrogen receptor (tamoxifen) are mainstays of treatment for patients with ER (estrogen receptor) and PR (progesterone receptor) positive breast cancer. However, patients relapse subsequent to treatment with these agents.
When estradiol binds to the ER, the MEK and Akt/PKB signal transduction pathways are activated, which drives the cancer. The ER acts as a nuclear transcription factor, which potentiates the transcription of genes driving proliferation and apoptosis inhibition (see diagram from Clinical Cancer Research).
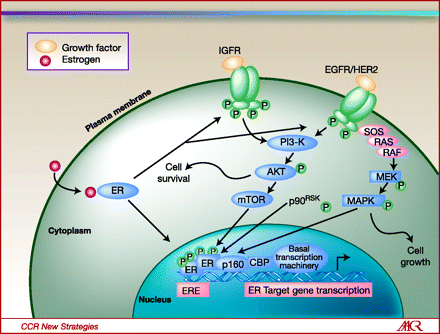
Cross-talk between signal transduction pathways and ER signaling in endocrine-resistant breast cancer, with opportunities for targeted intervention. Estrogen (E2)-liganded ER activates E2-regulated genes in classical pathway, but following long-term endocrine therapy, resistance can develop with bidirectional cross-talk between ER and growth factor receptors, with association of membrane-bound ER with growth factor receptors, and/or IGFR or EGFR/HER2 activation of ER phosphorylation. Modulating these pathways by various signal transduction inhibitors may overcome the resistance to endocrine therapy.
SERDs are designed to not only block estradiol action at the estrogen receptor, but eliminate the ER by changing its conformation such that the cell targets the ER for destruction. This would provide a much more efficient and complete blockade of the ER. Genentech paid $725MM up front, with a potential 1$B more based on achievements of certain milestone. ARN-810 is in early clinical studies (Phase 1), which is why this sort of deal stands-out. Normally, a big company like Genentech would license the product at this stage, but here, they bought the whole company. Why?
Because, Seragon is a company that has a very successful history of developing first in class products for hormone-driven diseases. Case in point, Zytiga for prostate cancer, which was acquired by J&J when it bought Aragon Pharmaceuticals; Seragon was spun-out from Aragon in the J&J acquisition.
Zytiga, abiraterone acetate, is converted in vivo to abiraterone, an androgen biosynthesis inhibitor, that inhibits 17 α-hydroxylase/C17,20-lyase (CYP17). This enzyme is expressed in testicular, adrenal, and prostatic tumor tissues and is required for androgen biosynthesis. CYP17 catalyzes two sequential reactions: 1) the conversion of pregnenolone and progesterone to their 17α-hydroxy derivatives by 17α-hydroxylase activity and 2) the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione, respectively, by C17, 20 lyase activity. DHEA and androstenedione are androgens and are precursors of testosterone. Inhibition of CYP17 by abiraterone can also result in increased mineralocorticoid production by the adrenals.
The AR is a nuclear receptor transcription factor; upon binding with DHT (dihydroxytestoerone), it translocates to the nucleus and induces the transcription of genes that promote growth and survival (like the ER). See diagram in BOLO.

Androgen Receptor activation and translocation to nucleus
The mainstay of treatment for prostate cancer is androgen deprivation, which can be accomplished with surgery (orchiectomy) or drugs that block the hypothalamic/pituitary axis, and the androgen receptor (AR).
With Zytiga, androgen production, itself is blocked, which is a more efficient way to stop androgen/AR interactions for many reasons. First, in castration-resistant prostate cancer, the cancer itself begins to produce testosterone (autocrine growth) and the testosterone receptors become much more sensitive to even low levels of androgen. Further, even low levels of testosterone that result from adrenal gland production can drive the tumor. So, blocking the biosynthesis of testosterone, as opposed to the signals for its production or its receptor binding is a much more efficient and ubiquitous way to achieve androgen deprivation therapy (see diagram from Nature Reviews).

Methods of achieving androgen deprivation for prostate cancer