
MET is a gene that encodes a receptor tyrosine kinase that is activated upon binding with hepatocyte growth factor (HGF, or Scatter Factor). Specifically, MET is a
Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis (By similarity). Acts as a receptor for Listeria internalin inlB, mediating entry of the pathogen into cells.
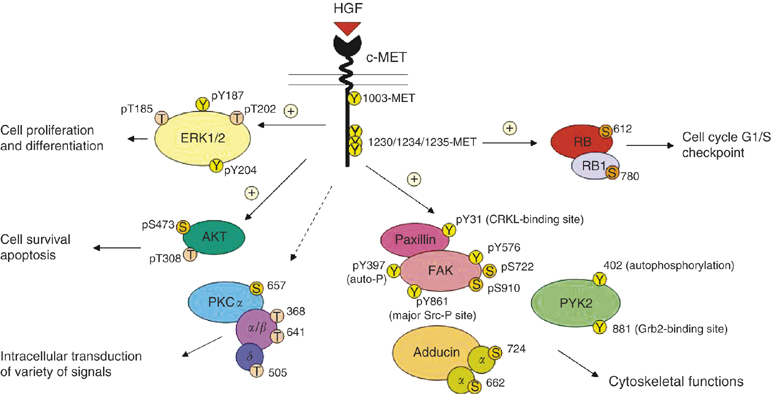
Figure 1. MET/HGF signal transduction pathways in SCLC. A schematic diagram to illustrate the versatile signalling functions of c-MET/HGF pathway in SCLC regulating various biological functions of the cells, including cytoskeletal functions, cell proliferation and differentiation, survival, and apoptosis is shown. Various potential serine, threonine and tyrosine phosphorylation sites on the signalling phosphoprotein intermediates are included. =stimulatory; =inhibitory. https://www.researchgate.net/publication/6171237_Downstream_signalling_and_specific_inhibition_of_c-METHGF_pathway_in_small_cell_lung_cancer_Implications_for_tumour_invasion
MET/HGF in epithelial-mesenchymal transition
MET binding of HGF induces the EMT (epithelial-mesenchymal transition), whereby epithelial cells acquire traits of mesenchymal cells, including E-cadherin to N-cadherin switching, anchorage independent growth, and invasiveness. This is a late transformation that carcinomas must undergo in order to metastasize.
HGF, EGF, PDGF, and TGF-β, appear to be responsible for the induction or functional activation in cancer cells of a series of EMT-inducing transcription factors, notably Snail, Slug, zinc finger E-box binding homeobox 1 (ZEB1), Twist, Goosecoid, and FOXC2 (66, 71, 76–79). Once expressed and activated, each of these transcription factors can act pleiotropically to choreograph the complex EMT program, more often than not with the help of other members of this cohort of transcription factors. The actual implementation by these cells of their EMT program depends on a series of intracellular signaling networks involving, among other signal-transducing proteins, ERK, MAPK, PI3K, Akt, Smads, RhoB, β-catenin, lymphoid enhancer binding factor (LEF), Ras, and c-Fos as well as cell surface proteins such as β4 integrins, α5β1 integrin, and αVβ6 integrin (80). Activation of EMT programs is also facilitated by the disruption of cell-cell adherens junctions and the cell-ECM adhesions mediated by integrins.
MET/HGF in resistance to targeted therapeutic approaches
Deregulation of MET/HGF (for example, amplification and constitutive firing) is responsible for resistance to targeted therapeutic strategies against, for example Braf, EGFR, and VEGF.
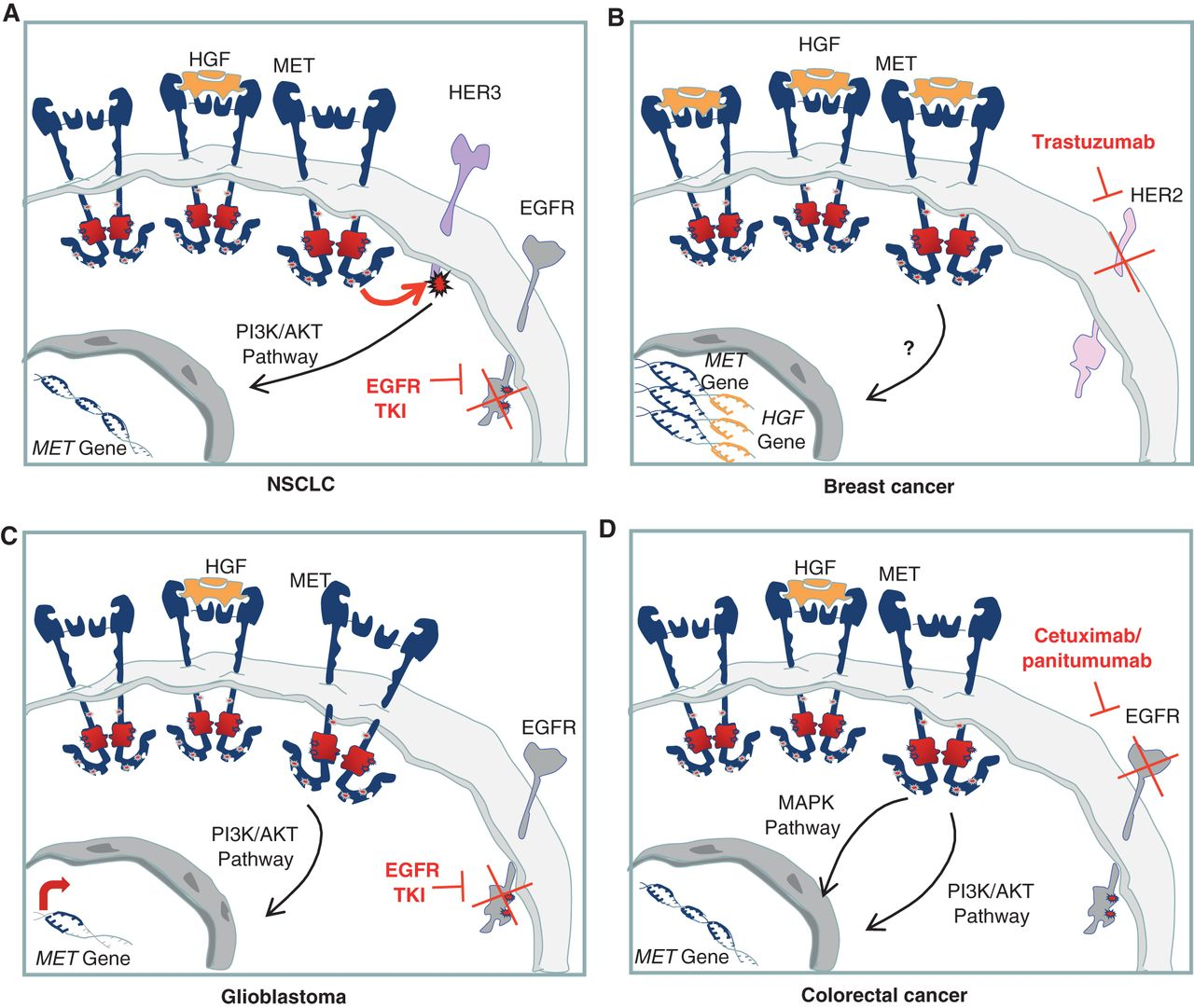
Figure 2. Cell-autonomous mechanisms of resistance to targeted therapies involving the HGF/MET axis. A, MET gene amplification results in overexpression of the receptor and HER3 transphosphorylation in NSCLC cells in which EGFR is inhibited by specific TKIs. B, chromosome 7 duplication results in increased copy number of both HGF and MET and, thus, in increased MET-dependent signal transduction. This results in resistance to trastuzumab in breast cancer cells. C, upon EGFR inhibition, a substantial increase in MET expression is responsible for sustained prosurvival AKT signaling, leading to resistance to EGFR TKIs in glioblastoma. D, MET gene amplification, causing receptor overexpression and activation of downstream pathways, induces resistance to cetuximab and panitumumab in colorectal cancer. http://cancerdiscovery.aacrjournals.org/content/3/9/978.figures-only

Figure 3. Non–cell-autonomous mechanisms of resistance to targeted therapies involving the HGF/MET axis. A, HGF secretion by stromal cells results in MET activation and stimulation of PI3K/AKT and MAPK pathways in tumor cells harboring BRAF-activating mutations or constitutive RTK activation (cancer cell lines with constitutive EGFR activation, constitutive HER2 activation, amplified FGFR, amplified PDGFR, ALK translocation or mutation; NSCLC, colon, gastric, and breast cancer). This leads to resistance to inhibitors targeting BRAF or growth factor receptors, respectively. B, HGF production by stromal cells can contribute to resistance to treatment with the antiangiogenic drug sunitinib by activating MET-dependent pathways in endothelial cells that compensate for VEGFR inhibition. http://cancerdiscovery.aacrjournals.org/content/3/9/978.figures-only
Anti-MET Therapy
Given the criticality of the MET/HGF axis in the induction of the EMT and in the emergence of resistance to targeted therapies, anti-MET kinase targeted therapy is a sound strategy. However, to date, this approach has not led to clinical success.
That is why AbbVie and BMS are joining forces in a clinical trial of the former’s antibody drug conjugate that targets MET combined with checkpoint inhibitor, novulmab, in patients with non-small cell lung cancer.
Telisotuzumab vedotin is a first-in-class anti-c-Met antibody drug conjugate (ADC) that targets both MET-amplified and c-Met-overexpressing tumors. It is currently being investigated to treat advanced solid tumors. c-Met expression is significantly higher in many solid tumors compared to normal tissue and is a marker of poor prognosis.
The MET receptor undergoes internalization following binding of its ligand. ADC’s exploit this biology – upon internalization, the antibody-receptor complex is taken up by lysosomes where vedotin, a potent microtubule toxin, is cleaved from the ABT-700 molecule to which it is joined via a protease-cleavabe linker. Vedotin leads to cell-cycle arrest and apoptosis.
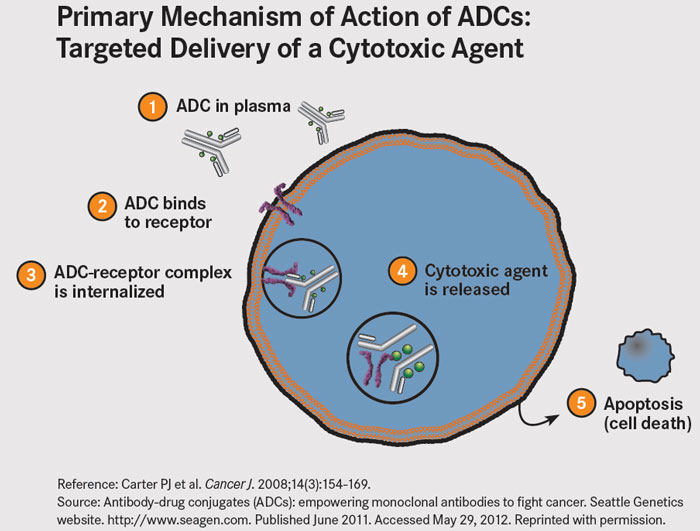
Figure 4. Reference: Carter PJ et al. Cancer J. 2008;14(3):154-169.
Source: Antibody-drug conjugates (ADCs): empowering monoclonal antibodies to fight cancer. Seattle Genetics website. seagen.com. Published June 2011. Accessed May 29, 2012. http://www.onclive.com/publications/oncology-live/2012/june-2012/antibody-drug-conjugates-guided-missiles-deployed-against-cancerous-cells
Although prior efforts at targeting c-Met overexpression have met with limited clinical success, an ADC directed against c-Met represents a unique strategy to deliver a potent cytotoxin directly to c-Met+ tumor cells.
ABBV-399 inhibits growth of xenograft tumors refractory to other c-Met inhibitors and provides significant therapeutic benefit in combination with standard of care chemotherapy. Antitumor activity was evaluated in cancer cells with overexpressed c-Met or amplified MET and in xenografts including patient-derived xenograft (PDX) models and those refractory to other c-Met inhibitors. Anti-c-Met monoclonal antibody ABT-700 binds to c-Met, thereby preventing c-Met binding to its ligand, HGF and the subsequent activation of the HGF/c-Met signaling pathway. This may cause cell death in c-Met-expressing tumor cells. c-Met, a receptor tyrosine kinase overexpressed or mutated in many tumor cell types, plays a key role in cancer cell growth, survival, angiogenesis, invasion, and metastasis.
A Phase 1/1b Study With ABBV-399, an Antibody Drug Conjugate, in Subjects With Advanced Solid Cancer Tumors is ongoing; it will include 292 patients. Cohort A is the dose escalation phase during which ABBV-399 will be administered, alone. Once the maximum tolerated dose is established, additional cohorts will evaluate ABBV-399 in combination with other treatments:
- Cohort B: ABBV-399 + erlotinib (an EGFR tyrosine kinase inhibitor)
- Cohort C: ABBV-399 + cetuximab (anti-EGFR monoclonal antibody)
- Cohort D: ABBV-399 + bevacizumab (anti-TNF monoclonal antibody)
- Cohort E: ABBV-399 + nivolumab