
Sitravatinib (MGCD516) is an oral multi-tyrosine kinase inhibitor being developed by Mirati Therapeutics. Last week, the company announced that three of eleven patients with non-small cell lung cancer (NSCLC) with genetic alterations in MET, AXL, RET, TRK, DDR2, KDR, PDGFRA, KIT or CBL who were resistant to checkpoint [anti PD-(L)1 therapy] had confirmed partial responses; because of this, dosing in the 34-patient expansion cohort will proceed.
Sitravatinib selectively targets receptor tyrosine kinases (RTKs) including TAM receptors (Tyro, Axl, MER), split family receptors (VEGFR2, KIT), RET and MET. These represent about 9% of NSCLC:

Figure 1. Genetic alterations in NSCLC targeted by sitravatinib. https://www.mirati.com/mgcd516/
In addition to targeting the driver mutations of the cancer, itself, these targets may enhance the antitumor response via direct effects on immune cells within the tumor microenvironment (TME):
- Inhibition of these target classes by sitravatinib may enhance anti-tumor activity through targeted depletion of immunosuppressive Type 2 tumor associated macrophages, regulatory T cells and myeloid-derived suppressor cells (MDSCs) and increasing antigen presentation capacity of dendritic cells in the TME.
- Targeting MERTK and Axl induces M1 macrophage responses, as opposed to immune-suppressive M2 responses.
- Targeting split receptors reduces Treg and MSDC (myeloid-derived suppressor cells) and releases brakes on the expansion of CD8+ cells induced by PD-1 inhibition.
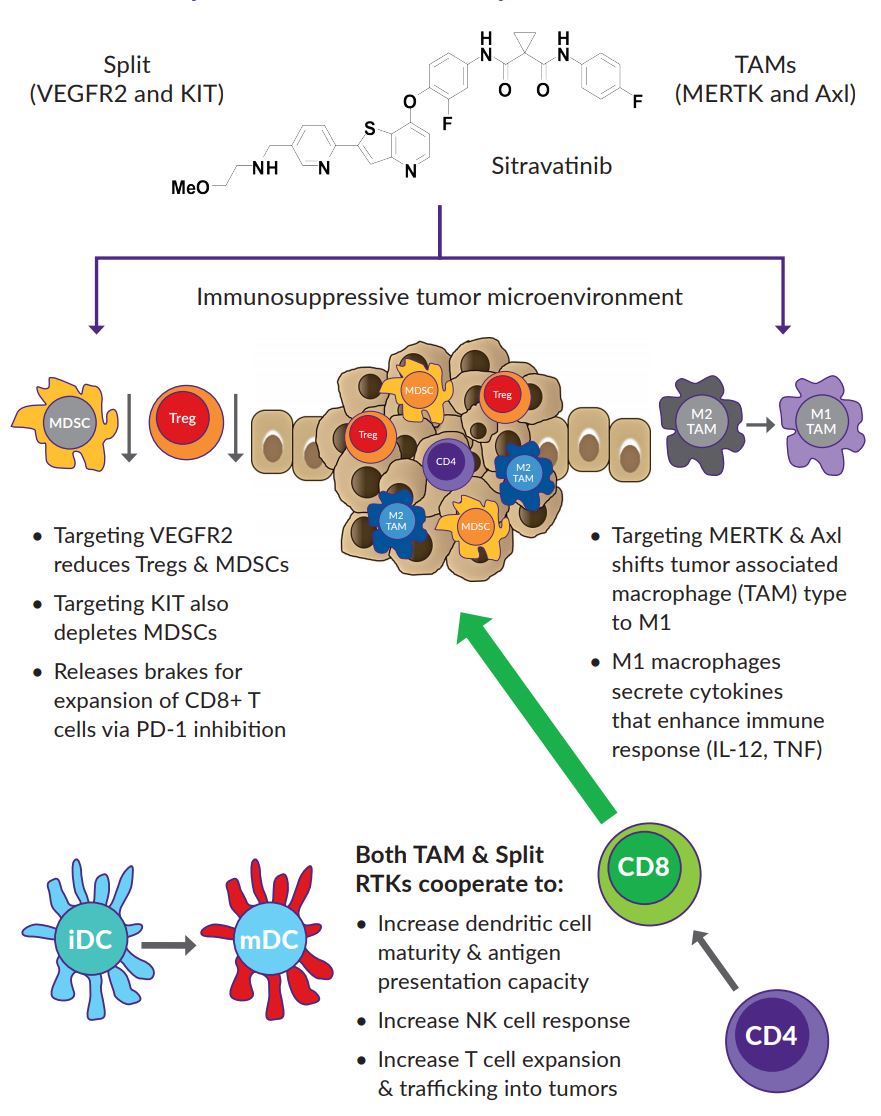
Figure 2. Rationale for Targeting TAM and Split RTKs to Enhance Response to Immune Checkpoint Inhibitors. https://www.mirati.com/wp-content/uploads/2017/09/AB4795_IASLC-Chicago2017_MRTX-500_SitravatinibCombo.pdf
How can sitravatinib turn “cold tumors hot?”
Sitravatinib is being developed in patients who have progressed on checkpoint therapy. These tumors no longer have activated infiltrating T-cells that are launching an effective immune attack. This is why checkpoint inhibition therapy is not working – if there is no anti-tumor immune activity ongoing, then, blocking the immune-abrogating signal (PD-L1) will not result in tumor kill.
But, PD-L1 expression is not the only reason that tumors escape immune attack – the presence of Treg cells that blunt CD8+ antigen specific reactions, MDSC’s, and M2 macrophage activity also contribute to making the tumor microenvironment immunologically cold.
So, the goal of treatment with sitravatinib in combination with Opdivo is several-fold:
- Sitravatinib therapy will kill many cancer cells that are driven by mutations in the tyrosine kinases that sitravatinib inhibits. This will result in the spilling of neo-antigens, epitopes against which the immune system has yet to launch an attack and to which the immune system has yet to be tolerized (Treg cells).
- Once an immune response against neo-antigens is initiated, Opdivo will block PD-L1-miedated immune response abrogation.
- Sitravatinib will also block the activation of Treg’s, promote M1 macrophage responses, and deplete the tumor microenvironment of MSDC’s.
Rationale for targeting CBL mutations with sitravatinib
CBL is a gene that encodes an E3 ubiquitin ligase, which is an enzyme that targets substrates for degradation by the proteasome. It mediates the transfer of ubiquitin from ubiquitin conjugating enzymes (E2) to specific substrates:
- CBL also contains an N-terminal phosphotyrosine binding domain that allows it to interact with numerous tyrosine-phosphorylated substrates and target them for proteasome degradation. As such it functions as a negative regulator of many signal transduction pathways. This gene has been found to be mutated or translocated in many cancers including acute myeloid leukaemia.
Loss of function mutations in CBL result in increased target RTK activation in tumor cells and may act as oncogenic drivers. CBL is commonly inactivated by missense mutations in the RING domain or by deletions that occur in selected human cancers including NSCLC, melanoma, and sarcomas.
CBL is negative regulator for MET, AXL, PDGFR/KIT signaling, therefore, blocking the tyrosine kinase activity of these RTKs with sitravatinib will address the loss of function of the CBL tumor suppressor gene when it is mutated.
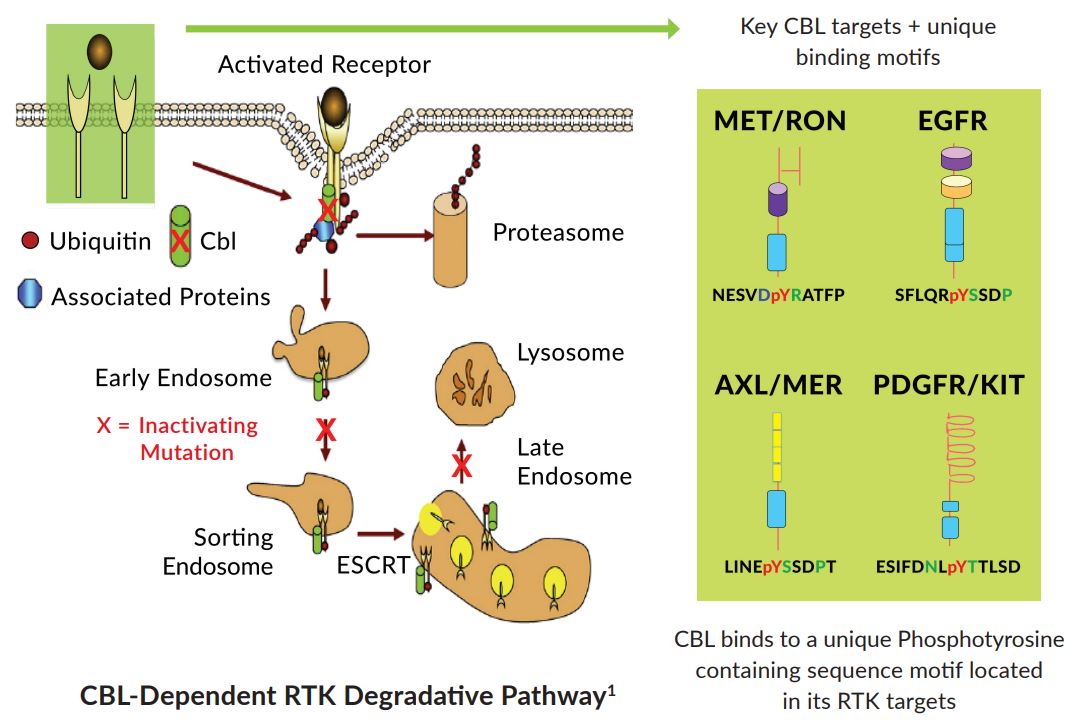
Figure 3. CBL degradation pathway of receptor tyrosine kinases. https://www.mirati.com/wp-content/uploads/2017/09/AB78_IASLC-Chicago_MGCD-516_Sitravatinib_CBL.pdf
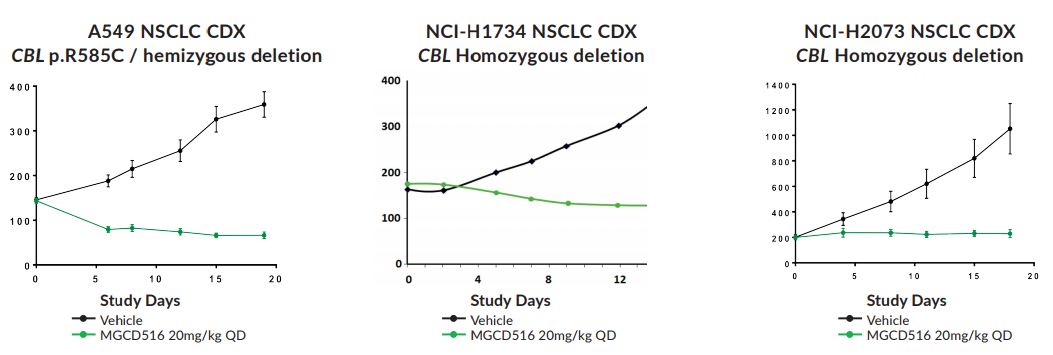
Figure 4. Sensitivity of NSCLC cell lines harboring CBL mutations to sitravatinib. https://www.mirati.com/wp-content/uploads/2017/09/AB78_IASLC-Chicago_MGCD-516_Sitravatinib_CBL.pdf