
CLEC12 (C-Type Lectin Domain Family 12 Member A) is negative regulator of granulocyte and monocyte functioning. It is a member of the C-type lectin/C-type lectin-like domain (CTL/CTLD) superfamily. It is also known as Myeloid Inhibitory C-Type Lectin-Like Receptor and Dendritic Cell-Associated Lectin. CLEC12 is a cell surface receptor that modulates signaling cascades and mediates tyrosine phosphorylation of target MAP kinases.
CLEC12A contains an ITIM (immune tyrosine-based inhibitory motif) that recruits the tyrosine phosphatases SHP-1 and SHP-2 to negatively regulate Syk signaling, a key pathway that drives inflammation through the production of reactive oxygen species (ROS) and generation of cytokines and chemokines. The ligand for CLEC12A is monosodium urate (MSU), which is released by dying cells. The function of CLEC12A, then, is to counterbalance the immune response to dead cells.
MSU activates proinflammatory responses not only by triggering NLRP3 but also by directly activating Syk at the cell surface of innate immune cells through ITAM-coupled CD11b/CD16 on neutrophils or induced lipid sorting and the subsequent formation of cholesterol rich regions that recruit ITAM-containing transmembrane adapters on dendritic cells (Barabé et al., 1998, Ng et al., 2008). In vivo, MSU is also found opsonized by complement components and antibodies, which can trigger Syk-coupled complement receptors and Fc receptors, respectively (Cherian and Schumacher, 1986, Terkeltaub et al., 1983). Our results demonstrating that Clec12a inhibits Syk-dependent ROS (reactive oxygen species) production induced by MSU suggest that Clec12a can counterbalance these Syk activating ITAM (immune tyrosine-based activation motif) signals. Consistent with this finding and our discovery that Clec12a directly binds MSU crystals, a current independent study reported enhanced tyrosine phosphorylation, Ca2+ signaling and IL-8 production upon MSU stimulation in a human neutrophil cell line after siRNA knockdown of Clec12a (Gagné et al., 2013). Similarly, downregulation of Clec12a with a monoclonal antibody enhanced the MSU-induced activation of human neutrophils. We found that this antibody (clone 50C1) also reduces Clec12a binding to MSU crystals (Figure S1), indicating that this antibody enhances neutrophil responses not merely by downregulating Clec12a but directly interferes with Clec12a ligand binding.
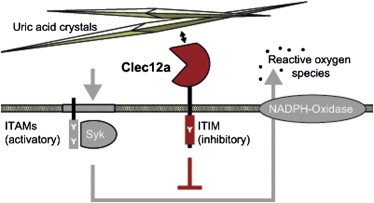
Figure 1. CLEC12A inhibits Syk signaling induced by uric acid crystals formed by dying cells. http://www.cell.com/immunity/fulltext/S1074-7613(14)00069-7
Where is CLEC12A expressed?
CLEC12A is expressed predominantly in the bone marrow and immune system (spleen, bone marrow, lymph nodes), as well as in the lung.
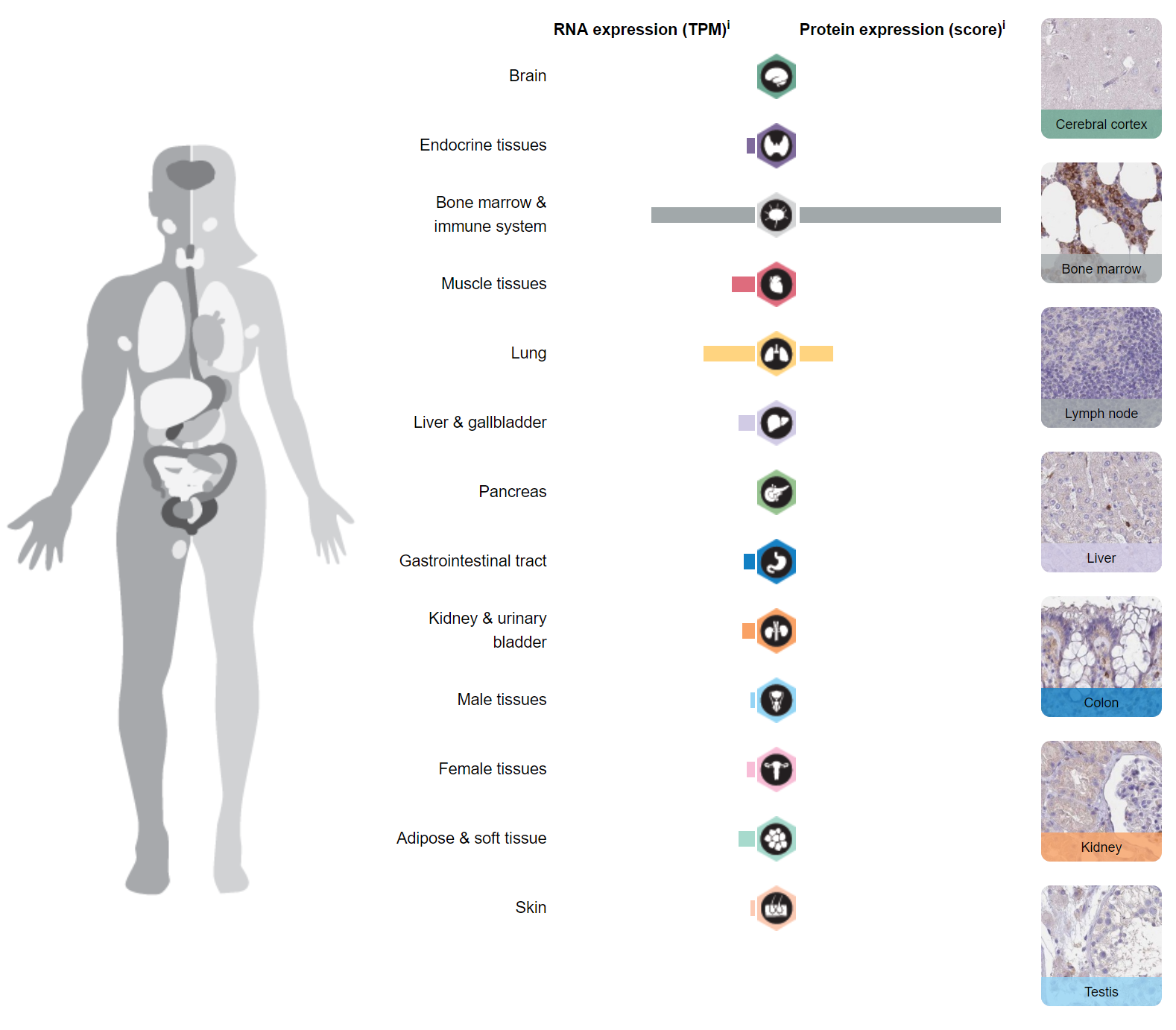
Figure 2. Tissue expression of CLEC12A. https://www.proteinatlas.org/ENSG00000172322-CLEC12A/tissue
CLEC12A in AML (Acute Myelogenous leukemia) and MDS (Myelodysplastic Syndrome)
CLECI12A is a leukemia‐associated marker that can be used diagnosis and to assess the presence of microscopic disease following ablative chemotherapy (minimal residual disease) in AML. It is a good marker for leukemic stem cells (LSC) since it can be found on CD34+CD38− cells in CD34‐positive AML, but is completely absent on the CD34+CD38− cells in normal donors and in regenerating bone marrow:
In a recent study, CLEC12A was aberrantly expressed on the CD34+ CD38− cell compartment in 71% (22/31) of MDS patients, distributed across all Revised International Prognostic Scoring System risk groups. The CD34+ CD38− CLEC12A+ cells were indeed malignant and possessed functional stem cell properties in the long‐term colony‐initiating cell assay. As opposed to reported findings in AML, cancer stem cells from MDS samples derived from both CLEC12A positive and negative CD34+ CD38- subpopulations.
CLEC12A positive blasts selected from AML patients were more resistant to chemotherapy compared to CLEC12A negative blasts (20% killing of CLEC12A positive AML cells versus 43% of CLEC12A negative AML cells when cultured with cytarabine 10 µg/ml, P=0.01). This finding was confirmed by using the AML MOLM14 cell line engineered to overexpress CLEC12A. CLEC12Ahigh MOLM14 cells were more resistant to chemotherapy compared to wild type MOLM14 cells (P=0.003). We then evaluated CLEC12A resistance to chemotherapy in a patient derived AML xenograft model. We found a relative increase in CLEC12A positive cells post Ara-C induction chemotherapy in AML xenograft.
Targeting CLEC12A therapeutically
Investigators at UPENN developed a chimeric T-cell against CLEC12A containing CD3z and 41BB costimulatory domains and generated CLEC12A CART cells by lentiviral transduction.
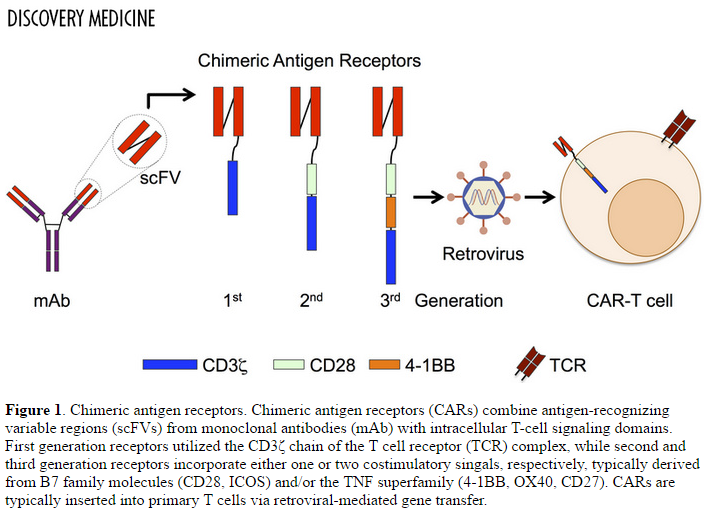
Figure 3. Chimeric antigen receptors. Chimeric antigen receptors (CARs) combine antigen-recognizing variable regions (scFVs) from monoclonal antibodies (mAb) with intracellular T-cell signaling domains. First generation receptors utilized the CD3ζ chain of the T cell receptor (TCR) complex, while second and third generation receptors incorporate either one or two costimulatory singals, respectively, typically derived from B7 family molecules (CD28, ICOS) and/or the TNF superfamily (4-1BB, OX40, CD27). CARs are typically inserted into primary T cells via retroviral-mediated gene transfer. http://www.discoverymedicine.com/Michael-S-Magee/2014/11/challenges-to-chimeric-antigen-receptor-car-t-cell-therapy-for-cancer/
The CAR T construct provided only modest results when used as monotherapy in AML xenografts. However, considerable activity was demonstrated when using the CAR T construct following chemotherapy, since chemotherapy destroys non-CLEC12A-expressing cells, enriching CLEC12A+ cells in minimal residual disease:
To investigate the potential role of CLEC12A CART cells in eradication of MRD and LSC, mice were treated first with chemotherapy (cytarabine 60 mg/kg intraperitoneal injection daily for 5 days) followed by a single dose (1×105 total T cells via tail vein injection) of either CLEC12A CARTs or control untransduced T cells (UTD). Treatment with CLEC12A CART cells resulted in eradication of leukemia and prolonged survival in these mice (overall survival at 200 days of 100% after CLEC12A CARTs compared to 20% after UTD, p=0.01).
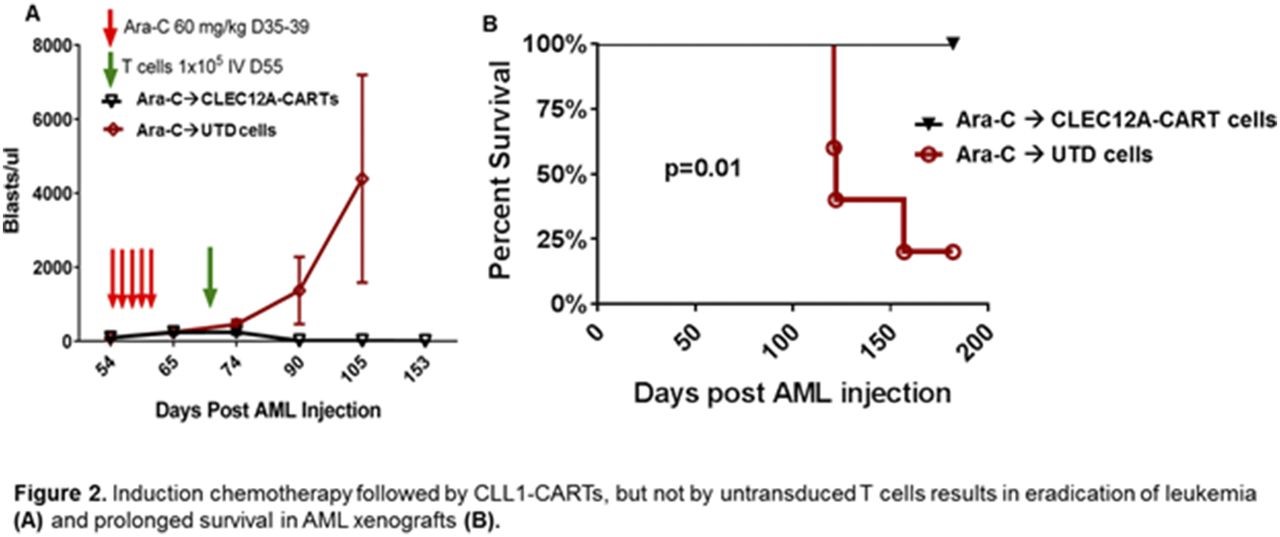
Figure 4. Induction chemotherapy followed by CLEC12A CAR T-cell treatment, but not by untransduced T-cells (UTD) results in eradication of leukemia and prolonged survival in AML xenografts. http://www.bloodjournal.org/content/128/22/766
Merus has used its “Biclonics” full-length IgG bispecific antibody platform to generate MCLA-117, which binds to CD3 (expressed on CD8+ cells) and CLEC12A. The approach is known as T-cell engagement, and is designed to localize activated T-cells to cells expressing CLEC12A, much in the same way that the bispecific antibody, Blincyto (blinatumomab), targets CD19 in ALL (acute lymphoblastic leukemia).
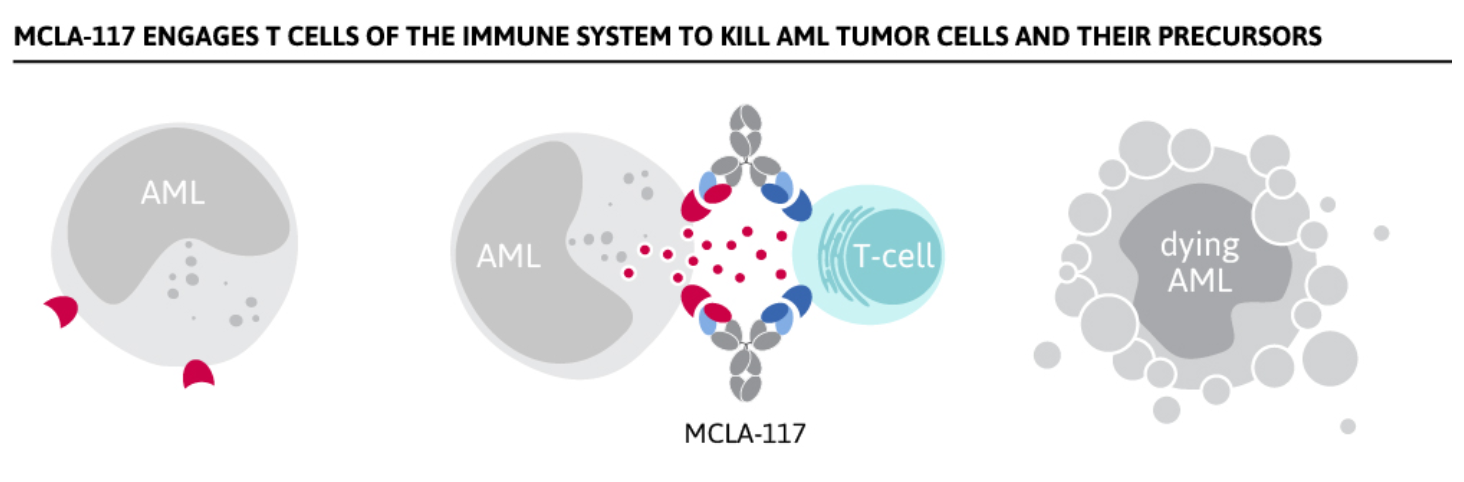
Figure 5. T-cell engagement to kill AML cells expressing CLEC12A. http://www.merus.nl/pipeline/
A first in man Phase 1 trial of MCLA-117 in patients with AML is underway in Europe. Patients must have at least 20% of cells expressing CLEC12A and satisfy the following requirements:
- Male or female age ≥18 years old;
- Signed informed consent form
- AML either de novo or secondary who either :
- are in relapse following an initial response and no more than 1 prior salvage therapy
- failed primary induction therapy with no complete remission and for whom no other approved therapy is available, and no more than 1 prior salvage therapy
- newly diagnosed untreated AML in patients ≥ 65 years of age, if they are not candidates for standard available induction chemotherapy