
Immune checkpoint-directed therapy is producing unprecedented clinical results in many patients. So much so, that the FDA recently reversed its longstanding policy or approving cancer drugs based on site of origin, to the presence of a biomarker (microsatellite instability (MSI-H) or mismatch-deficient repair (dMDR) as the indication for therapy with pembrolizumab (Ketruda), and PD-1 blocker. Cancers expressing MSI-H or dMDR mutate at a rapid rate, presenting novel epitopes to the immune system, which is readily mobilized against them so that tumor infiltrating T-cells are reliably present. Blocking the PD-1/PD-L1 pathway in this context allows for prolongation of the immune response and better clinical results.
For 15+ years, cancer experts have told us that with cancer, the most important issue is mutation, mutation, mutation, yet, the FDA approached the disease more consistent with the real estate adage, “location, location, location.” It took the amazing results with checkpoint inhibitors to move the FDA.
Stands to reason then, that many are pursuing the discovery and development of immune checkpoint inhibitors, as summarized in Table 1.
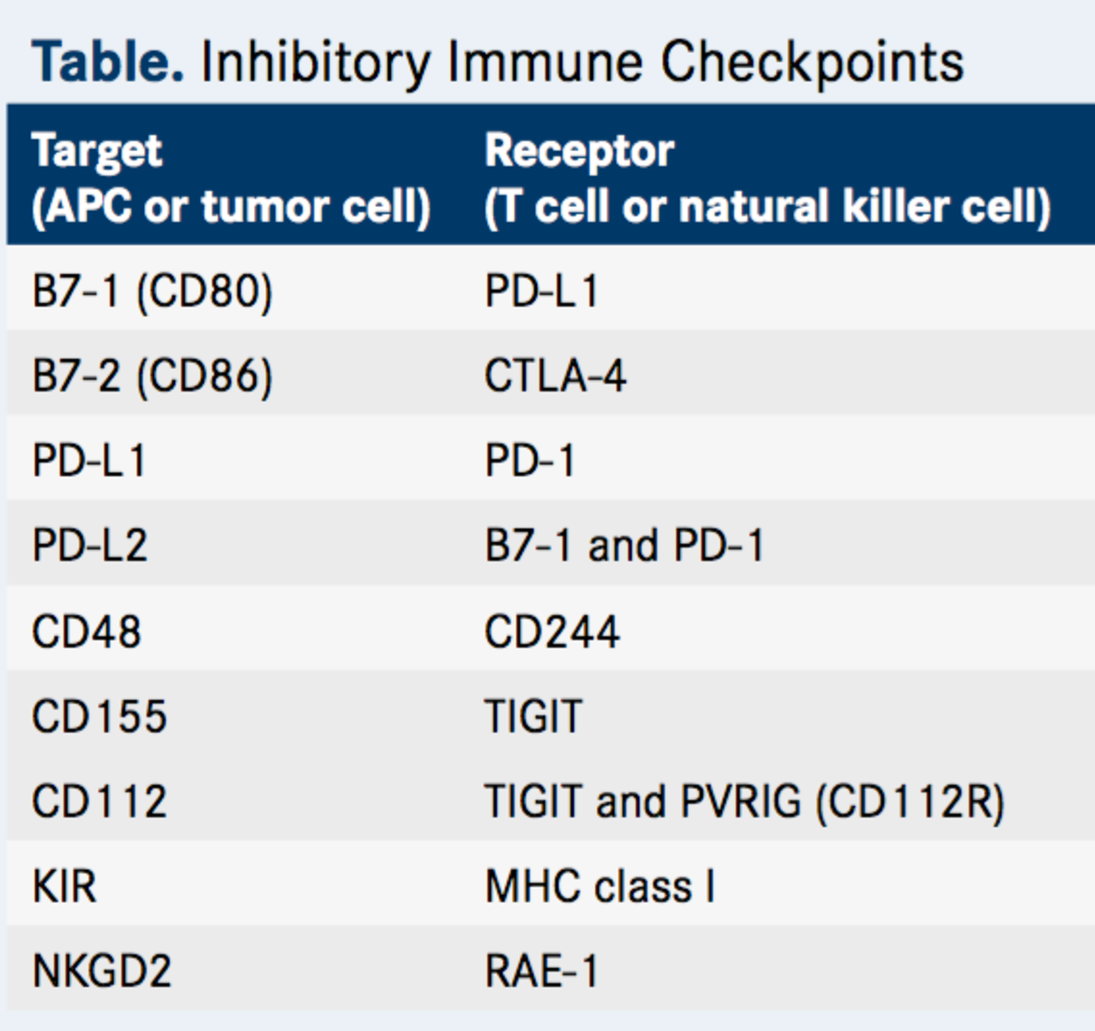
Table 1. Inhibitory immune checkpoints. http://www.onclive.com/publications/oncology-live/2017/vol-18-no-03/tigit-emerges-as-new-target-for-immune-checkpoint-blockade-strategies?p=2
One new target is TIGIT, an antibody-like poliovirus receptor protein present on T-cells and natural killer cells that contains immunoreceptor tyrosine-based inhibitory motifs (ITIM), which recruit tyrosine phosphatases that terminate signal transduction. [Immunoreceptor tyrosine-based activation motifs (ITAM), on the other hand, recruit tyrosine kinases to propagate signal transduction pathways. Immunoreceptor tyrosine-based switch motifs (ITSMs) recruit kinases for activation in some situations and phosphatases to inhibit signaling in others.]
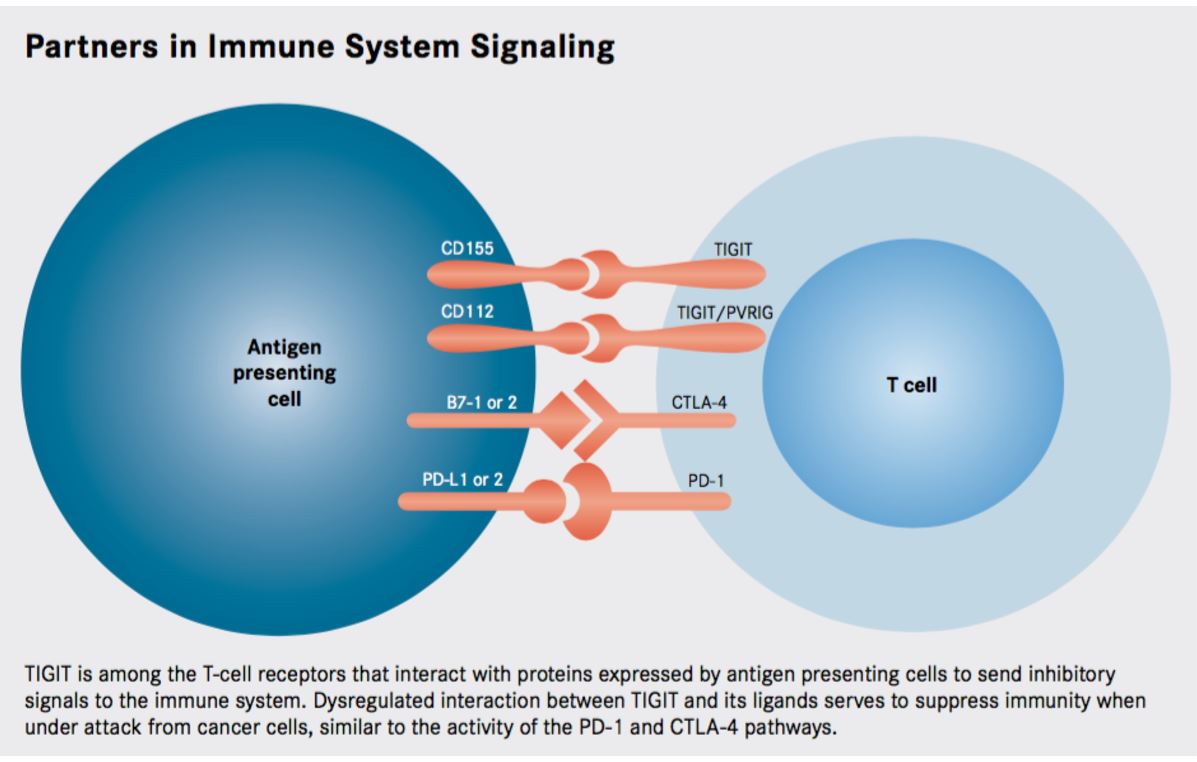
Figure 1. Researchers continue to identify a growing number of immune checkpoints as targets for anticancer therapy, the recently discovered TIGIT pathway is emerging as a promising new avenue for exploration. http://www.onclive.com/publications/oncology-live/2017/vol-18-no-03/tigit-emerges-as-new-target-for-immune-checkpoint-blockade-strategies#sthash.LcG57hTn.dpuf
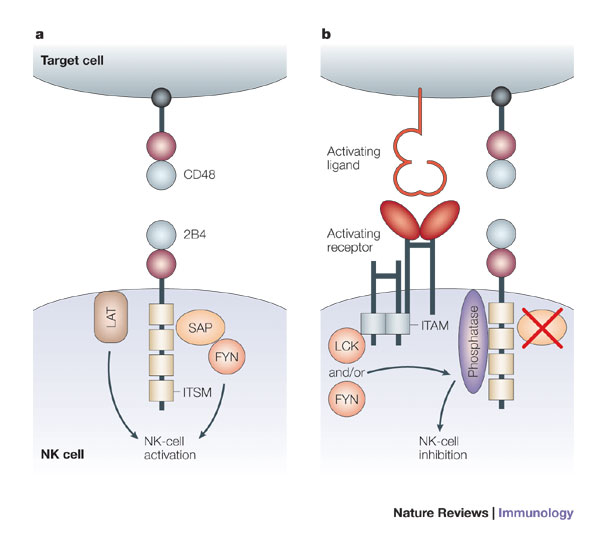
Figure 2. Immunoreceptor tyrosine-based switch motifs (ITSMs) are more versatile than traditional immunoreceptor tyrosine-based inhibitory motifs (ITIMs) and immunoreceptor tyrosine-based activation motifs (ITAMs), because ITSMs recruit kinases in some situations and phosphatases in others. a | Ligation of 2B4 leads to activation of mature human natural killer (NK) cells. When engaged by CD48, ITSMs in the cytoplasmic tail of 2B4 bind the SRC homology 2 (SH2) domain of SAP (signalling lymphocytic activation molecule (SLAM)-associated protein). SAP then recruits the kinase FYN, through the SH3 domain of FYN. Linker for activation of T cells (LAT) present in lipid-raft domains also associates with 2B4, and in conjunction with FYN, this interaction leads to the activation of NK cells. b | In mouse NK cells, as well as in NK cells from patients with X-linked lymphoproliferative syndrome (XLP) and immature human NK cells, 2B4 transmits inhibitory signals. In humans, this inhibition occurs in the absence of functional SAP, as occurs in patients with XLP and in immature NK cells. When 2B4 and an activating receptor are co-engaged by a potential target cell, the ITSMs of 2B4 are phosphorylated by the SRC-family kinases LCK and/or FYN. This leads to the recruitment of phosphatases, such as SHP1 (SH2-domain-containing protein tyrosine phosphatase 1) and SHP2, which in turn inhibit activation by dephosphorylating molecules downstream of the activating receptor. In mice, 2B4 transmits inhibitory signals regardless of the presence or absence of SAP. ITAM, immunoreceptor tyrosine-based activation motif. http://www.nature.com/nri/journal/v5/n5/fig_tab/nri1603_F3.html
How does TIGIT work?
TIGIT functions similarly to CTLA4, a T-cell co-stimulatory molecule, which when binding B7 (expressed on antigen presenting cells), emits negative signals within the T-cell, abrogating the immune response. B7 also binds to CD28 (a costimulatory molecule expressed on T-cells) to promote T-cell activation. Similarly, CD155, the ligand for TIGIT (abrogating signals), also binds to CD226 to provide activating/stimulatory signals to the T-cell.
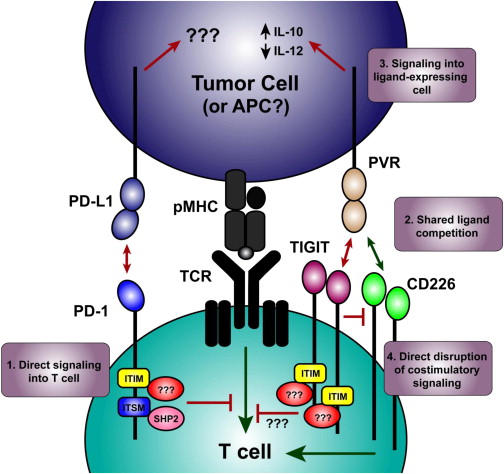
Figure 3 – TIGIT Signaling. Putative Molecular Mechanisms Involved in Synergy Observed during Coblockade. (1) Inhibitory receptors can deliver signals directly to the T cell by recruiting proteins to the intracellular domains (e.g., the ITSM of PD-1 recruiting SHP2). (2) Costimulatory and coinhibitory receptors that share ligands can compete for ligand binding (e.g., TIGIT and CD226 competing for PVR). (3) Inhibitory receptor binding can deliver an inhibitory signal to the ligand-expressing cells (e.g., TIGIT binding PVR induces functional changes in dendritic cells.) (4) Novel mechanism of inhibitory receptor function identified by Johnston et al. (2014)) where inhibitory receptors can directly block homodimerization of costimulatory receptors, mediating the inhibitory effect by preventing the costimulatory signal from acting (e.g., TIGIT physically preventing CD226 from signaling). Red lines indicate inhibitory signals, green lines indicate stimulatory signals. http://www.cell.com/cancer-cell/fulltext/S1535-6108(14)00467-X
TIGIT is reminiscent of the CTLA-4 protein, in that it shares ligands with an activating receptor. CTLA-4 is activated by binding to the B7-1 and B7-2 (also known as CD80 and CD86, respectively) proteins. These proteins also serve as ligands for the CD28 protein, a costimulatory molecule; thus the activating and deactivating receptors compete for the same ligand, with a delicate balance determining if the T cell is switched on or off.
The ligand for TIGIT is CD155 (alternatively known as PVR), but this protein also serves as a ligand for CD226, which, like CD28, is an activating receptor. When CD155 is bound to CD226, it conveys activating signals into the immune cell. Meanwhile, CD155 bound to TIGIT transmits an inhibitory signal by recruiting the SHP1 phosphatase to the membrane through its ITIM domain that subsequently deactivates numerous proteins involved in T-cell effector functions.
TIGIT is expressed by activated cytotoxic T cells and regulatory T cells and has also been shown to be upregulated on T cells in multiple cancer models. The ligands CD155 and CD112 are found on dendritic cells and macrophages and are also highly expressed in several types of cancer. Additionally, TIGIT expression is highly correlated with the expression of other coinhibitory molecules, including PD-1. Overall, this suggests that tumors upregulate the TIGIT pathway along with other inhibitory checkpoint networks to promote immunosuppressive mechanisms.
By binding to CD155 on the surface of dendritic cells, TIGIT increases the secretion of the immunosuppressive cytokine interleukin-10, and engagement of TIGIT on regulatory T cells enhances their immunosuppressive functions. TIGIT is also expressed on NK cells, the principal effector of the innate immune response that has been gaining traction as a target for anti-cancer therapy in recent years. Immune checkpoints have also been identified on NK cells, such as killer immunoglobulin-like receptor (KIR) that can be targeted to manipulate NK cell activity. In preclinical models, TIGIT blockade also boosts NK cell activation, suggesting that targeting TIGIT could offer a way of simultaneously boosting both arms of the immune response.
Most intriguingly, TIGIT has been shown to not simply outcompete CD226 for binding to CD155, but also to physically impede the dimerization of the activating receptor, blocking its costimulatory function. Thus, TIGIT inhibitors might not only release the brakes on the immune system, but at the same time may hit the gas by releasing TIGIT inhibition of CD226.
Who is working on TIGIT?
Genentech: Launched a phase 1 trial of MTIG7192A, which binds to TIGIT, combined with atezolizumab (Tecentriq, anti-PD-L1 antibody). It will recruit 300 patients.
Bristol Myers Squibb (BMS): Launched a phase 1/2 trial of BMS-986207, an anti-TIGIT monoclonal antibody, combined with nivolumab (Opdivo, anti-PD-1 antibody). It will include 170 patients.
OncoMed: Launched a phase 1 trial of OMP-31M32, a monoclonal antibody that binds to TGIT. Celgene entered into a deal with OncoMed for rights to this product – CELG will make a $35 million payment to OncoMed at the conclusion of the phase 1 trial; all future development costs would be paid by CELG (with up to $1.5 billion in potential payout, not including royalties).
Compugen: developing COM701, an antibody targeting PVRIG. An IND is planned for later in 2017.