
Checkpoint inhibitors, alone, are effective in 25% of patients when administered as a single agent. The goal of the collaboration between Celgene and Jounce is to address the other 75% of patients. The lead program is focused on ICOS, the Inducible T cell CO-Stimulator, a protein on the surface of T cells that can spur an immune response against a patient’s cancer.
Checkpoint Inhibition
ICOS stimulation differs from checkpoint inhibition in that it aims to stimulate a cytotoxic T-cell response, as opposed to blocking the signals (e.g., the PD-1/PD-L1 axis, Figure 1) that abrogate an immune attack on cancer cells. Celgene has a collaboration in place with AstraZeneca’s PD-L1 durvalumab.
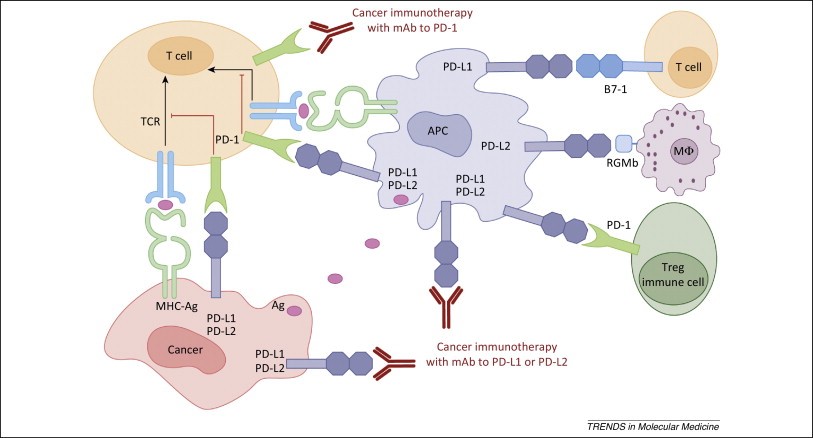
Figure 1. Human cancer immunotherapy with anti-PD-1 and anti-PD-L1/L2 antibodies. Antigen-presenting cells (APC) take up antigens (Ag) released from cancer cells and present to T cells. Cancer cells can also present Ag to activated T cells in the context of MHC. Upon T cell activation, PD-1 receptors are expressed on T cells and inhibit immune responses by engagement of PD-L1 and PD-L2 ligands on APC and PD-L1 on cancer cells. Therefore, monoclonal antibody (mAb)-mediated specific blockade of the PD-1/PD-L1/PD-L2 pathway can enhance anti-tumor immunity. In addition to binding to PD-1, PD-L1 and PD-L2 also bind B7-1 and repulsive guidance molecule B, respectively. In addition to T cells and APC, PD-1 and PD-L1 can be induced on other immune cells. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4282825/
PD-1 has two binding ligands, PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273), with PD-L1 being the most prominent in regulation. PD-L1 is inducibly expressed on both hematopoietic cells and non-hematopoietic cells following cell-specific stimulation. Cytokines such as IFN-γ and TNF-α up-regulate the expression of PD-L1 on T cells, B cells, endothelial cells, and epithelial cells, furthering its role in the maintenance of peripheral tolerance. Data also links genetic changes seen in cancer cells to the induction of PD-L1, although this can vary by cancer type. PTEN dysfunction in human glioma cells induces Akt activation and subsequently PD-L1 expression, while human melanoma cells show no association between PTEN or Akt and PD-L1 induction. Recent data shows that PD-L1 binds to B7-1 (CD80) in addition to PD-1. While PD-L1 expression is induced on a wide array of both hematopoietic and non-hematopoietic cells, PD-L2 expression is restricted to inducible expression on DCs, macrophages, mast cells, and some B cells in response to IL-4 and IFN. The affinity of PD-L2 for PD-1 is three times greater than that of PD-L1, which indicates competition between the two ligands. Recent data confirms a second cognate receptor for PD-L2, repulsive guidance molecule B (RGMb). Despite recent research efforts surrounding PD-L2, little is known regarding the transcriptional regulation of the ligand.
Other immune checkpoints include LAG3, 2B4 (CD244), killer cell lectin-like receptor subfamily G member 1 (KLRG1), CD160, B and T lymphocyte attenuator (BTLA) and T cell immunoglobulin domain and mucin domain 3 (TIM3) – Figure 2.
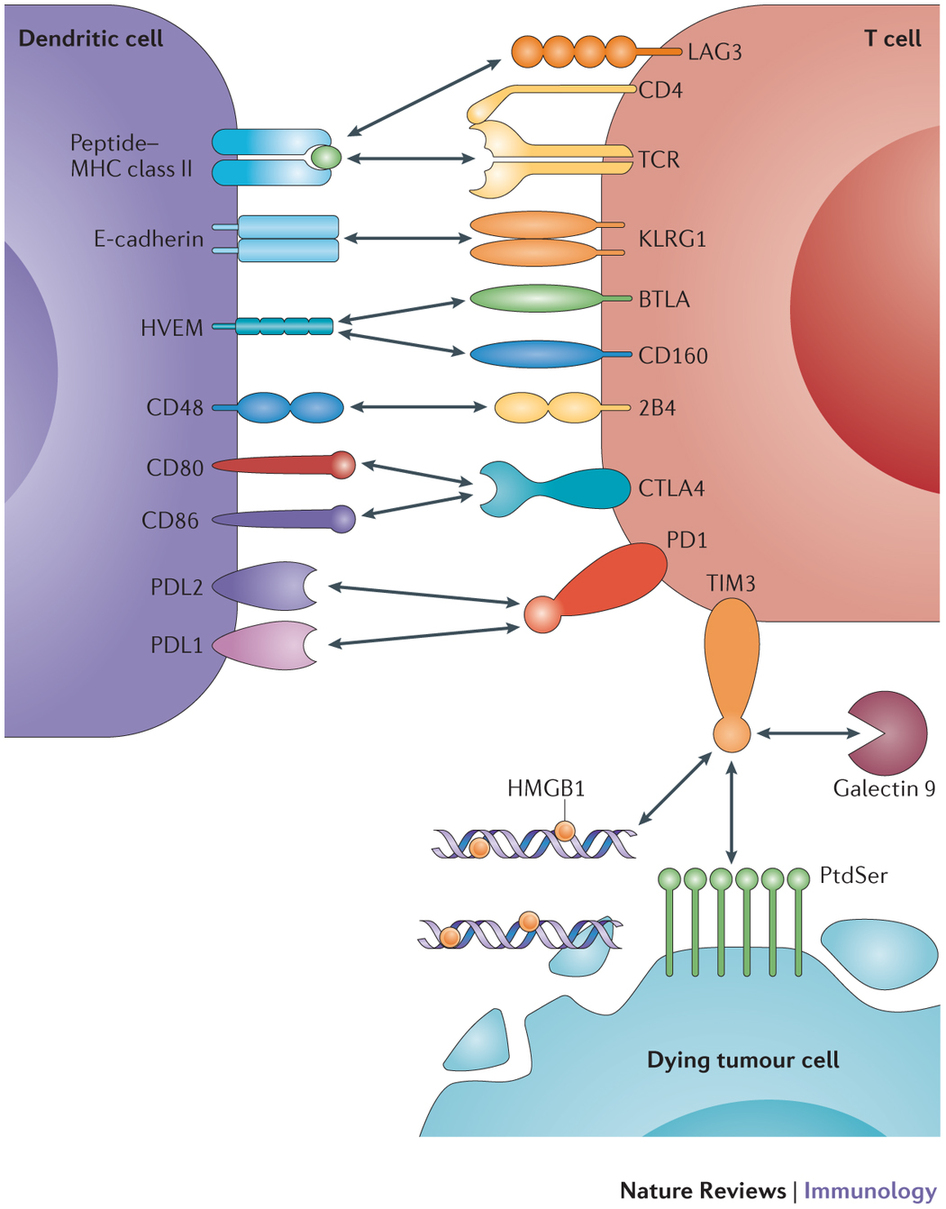
Figure 2. Inhibitory receptors and their ligands are depicted. The receptors include cytotoxic T lymphocyte antigen 4 (CTLA4), programmed cell death protein 1 (PD1), lymphocyte activation gene 3 protein (LAG3), 2B4 (also known as CD244), killer cell lectin-like receptor subfamily G member 1 (KLRG1), CD160, B and T lymphocyte attenuator (BTLA) and T cell immunoglobulin domain and mucin domain 3 (TIM3). Their ligands include CD80, CD86, programmed cell death protein 1 ligand 1 (PDL1), PDL2, MHC class II, CD48, E-cadherin, herpesvirus entry mediator (HVEM), galectin 9, phosphatidylserine (PtdSer) and high-mobility group box 1 protein (HMGB1). Only the interactions that mediate negative regulation are shown; some molecules have binding partners in addition to the ones shown that mediate other functions. In addition to the negative regulatory interactions, the interaction between the T cell receptor (TCR) and peptide–MHC class II is also shown as this is essential for T cell function. Note that dendritic cells also express MHC class I molecules and the negative regulatory interactions shown here can occur between dendritic cells and CD4+ T cells or CD8+ T cells. In this figure, PtdSer and HMGB1 are shown emanating from dying tumour cells. However, a stressed tumour cell could also upregulate these TIM3-binding partners. Bidirectional negative signalling between PDL1 and CD80 has also been described. http://www.nature.com/nri/journal/v15/n1/full/nri3790.html
The Inducible T-cell CO-Stimulator
JTX-2011 is an agonist monoclonal antibody to ICOS (The Inducible T-cell CO-Stimulator) that is in the pre-IND development stage. Elevated expression of ICOS correlated with positive clinical outcomes; providing an agonist signal through ICOS leads to anti-tumor activity. The protein encoded by this gene belongs to the CD28 and CTLA-4 cell-surface receptor family. It forms homodimers and plays an important role in cell-cell signaling, immune responses, and regulation of cell proliferation.
The T-cell-specific cell-surface receptors CD28 and CTLA-4 are important regulators of the immune system. CD28 potently enhances those T-cell functions that are essential for an effective antigen-specific immune response, and the homologous CTLA-4 counterbalances the CD28-mediated signals and thus prevents an otherwise fatal overstimulation of the lymphoid system. A third member of this family of molecules, the inducible co-stimulator (ICOS), is a homodimeric protein of relative molecular mass 55,000-60,000 (M(r) 55K-60K). Matching CD28 in potency, ICOS enhances all basic T-cell responses to a foreign antigen, namely proliferation, secretion of lymphokines, upregulation of molecules that mediate cell-cell interaction, and effective help for antibody secretion by B cells. Unlike the constitutively expressed CD28, ICOS has to be de novo induced on the T-cell surface, does not upregulate the production of interleukin-2, but superinduces the synthesis of interleukin-10, a B-cell-differentiation factor. In vivo, ICOS is highly expressed on tonsillar T cells, which are closely associated with B cells in the apical light zone of germinal centres, the site of terminal B-cell maturation. Our results indicate that ICOS is another major regulator of the adaptive immune system.
ICOS, a third member of the CD28/CTLA-4 family, is expressed on activated T cells, but very little on resting naïve T cells. ICOS binds specifically to its ligand (ICOS-L), also known as B7-related protein-1 (B7RP-1), which is expressed constitutively on B cells. In vivo, the interaction of ICOS with ICOS-L is critical for T cell-dependent B cell responses. In the absence of ICOS, germinal center formation is impaired and immunoglobulin class switching is defective. In addition to providing help for B cells, ICOS plays an important role in the differentiation of unpolarized CD4+ T cells into Th1, Th2, Th17, and Treg lineages. It has been shown that ICOS can promote both Th1 and Th2 responses. However, ICOS and ICOS-L interaction during the early stages of T cell differentiation favors Th2 response. Although ICOS is not required for Th17 differentiation during primary responses, it plays a pivotal role in promoting Th17 compartment by upregulating IL-23R during secondary responses. Recent studies also demonstrate a critical role of ICOS in maintaining the homeostasis of CD4 + Foxp3 + Treg.
T cell-dependent B cell responses are critically dependent on the interaction of ICOS with ICOS-L, which promotes germinal center formation and immunoglobulin class switching in vivo. It has been shown that anti-ICOS alone or in combination with CD40Ig or anti-CD40L can induce tolerance to islet allografts and inhibit allograft rejection in murine models of transplantation. In a murine model of non-viral mediated gene therapy for hemophilia A, blockade of the ICOS–ICOS-L pathway by anti-ICOS antibody alone was effective in inhibiting the formation of anti-factor VIII antibodies following plasmid DNA-mediated gene transfer. In this study, anti-ICOS treatment also resulted in elevation of Treg numbers and their suppressive activity.
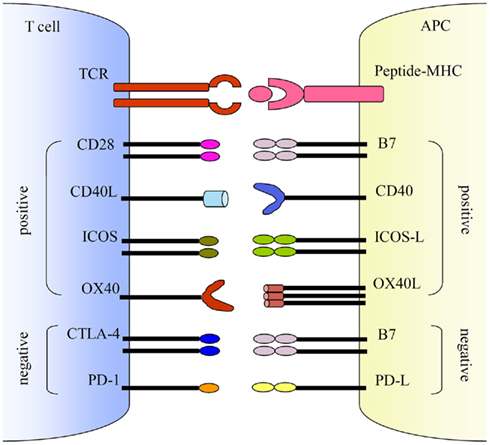
Figure 3. Co-stimulations either enhance or down-regulate T cell activation following the initial TCR and peptide-MHC ligation. Positive co-stimulatory pathways include B7–CD28, CD40L–CD40, ICOS–ICOS-L, and OX40–OX40L. Negative co-stimulatory pathways include B7–CTLA-4 and PD-1–PD-L. http://journal.frontiersin.org/article/10.3389/fmicb.2011.00202/full
Collaboration with Celgene
Celgene is paying $225 million in the upfront, adding $36 million in equity and committing up to $2.3 billion in milestones to complete the deal. In turn the big biotech gets dibs on a 40% share of U.S. profits for Jounce’s lead drug, the preclinical JTX-2011, plus a 75% share for its unnamed successor and a split on three more programs. There’s also an opt-in available on an experimental checkpoint program taking shape at Jounce. Once Celgene chooses to opt in, the two companies will divvy up expenses the same way they plan to share profits. And Celgene gets all ex-U.S. revenue with a royalty stream for Jounce.