

It is estimated that about 40 percent of patients with advanced melanoma, the deadliest form of skin cancer, will initially respond to an immunotherapy, but about a quarter of those 40 percent will relapse within three years of treatment. In order to identify the mechanisms by which resistance to PD-1 inhibition is mediated, UCLA researchers studied biopsies of melanoma tumors taken before and after treatment with Keytruda (pembrolizumab) in patients whose cancer had returned.
Their study was published in the New England Journal of Medicine and is summarized herein.
Of 78 patients with metastatic melanoma who were treated with the anti–PD-1 antibody pembrolizumab at the University of California, Los Angeles (UCLA), 42 had an objective response, of whom 15 went on to have disease progression. Four of these 15 patients met all three selection criteria for this analysis. First, they must have had an objective tumor response while participating in a clinical trial with single-agent pembrolizumab. Tumor responses were evaluated at 12 weeks and confirmed 4 weeks later, and patients were assessed by imaging every 12 weeks thereafter with the use of both the Response Evaluation Criteria in Solid Tumors (RECIST) and the immune-related response criteria. Second, patients had to have late acquired resistance, defined as in situ recurrence or new lesion development, despite continuous dosing, after more than 6 months of tumor response. Third, patients had to have adequate biopsy material for whole-exome sequencing at two time points: before the initiation of pembrolizumab therapy and after disease progression. The mean time to relapse for the four patients was 624 days (range, 419 to 888).
Patients 1 and 2 – JAK1 and JAK2 mutations
Tumors from patients 1 and 2 developed mutations in JAK1 and JAK2 after treatment with pembrolizumab. JAK1 and JAK2 are part of the interferon-receptor pathway, transmitting signals to the nucleus when cells bind IFN-g. Inactivating JAK mutations may result in a functional advantage for the progressive tumors because the lack of interferon signaling either decreased antigen presentation or allowed escape from interferon-induced inhibition of growth.
In the case of Patients 1 and 2, of 1173 and 240 nonsynonymous mutations (those that result in changes in amino acid sequences), respectively, originally identified in the baseline sample, 92.5% and 95.8% were also seen in the resistant tumor. The relapsing tumors also contained the same chromosomal loss-of-heterozygosity events as the baseline tumors, and all differences were due to further loss in the relapse samples.
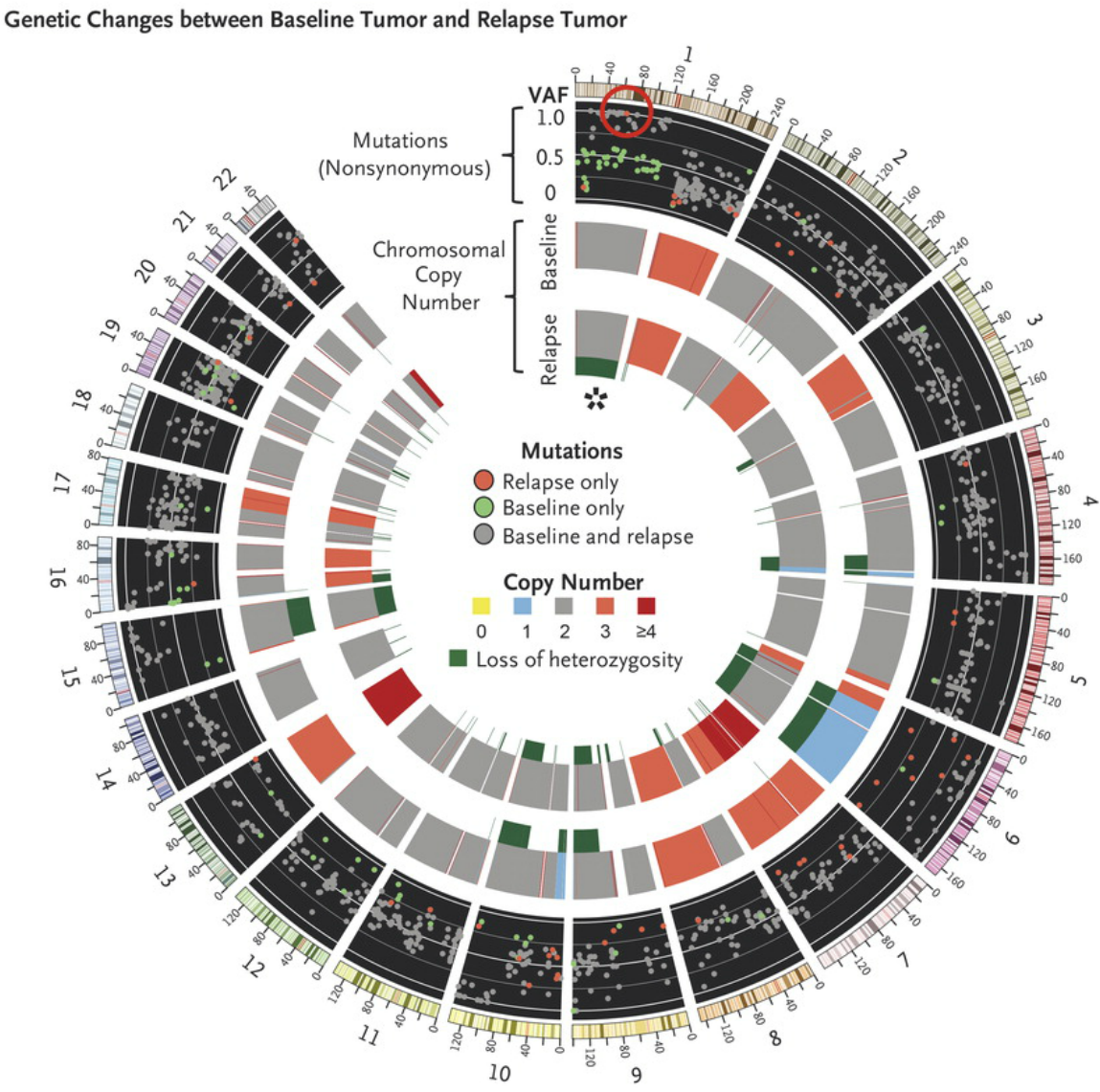
Differences in whole-exome sequencing between the pre-pembrolizumab and post-relapse biopsies. The red circle highlights a new, high-allele-frequency, relapse-specific mutation in the gene encoding Janus kinase 1 (JAK1) in the context of chromosomal loss of heterozygosity (asterisk). Each wedge represents a chromosome. In the outer track (black background), each point represents a nonsynonymous mutation, with most detected in both biopsy samples (gray) rather than at relapse only (red) or baseline only (green). The y-axis position indicates the variant allele frequency (VAF) at relapse, unless baseline-specific. The middle and inner tracks show copy-number status for the baseline and relapse biopsy, respectively; dark green in the subtrack indicates loss of heterozygosity. http://www.nejm.org/doi/full/10.1056/NEJMoa1604958#t=article
In the relapse biopsy samples from both patients, the researchers identified new homozygous loss-of-function mutations in the kinases associated with the interferon-receptor pathway, with a Q503* nonsense mutation in the gene encoding Janus kinase 1 (JAK1) in Patient 1 and a F547 splice-site mutation in the gene encoding Janus kinase 2 (JAK2) in Patient 2. RNA sequencing showed that the JAK2 splice-site mutation caused intron inclusion, producing an in-frame stop codon 10 bp after exon 12. Therefore, both mutations were upstream of the kinase domains and probably truncated the protein or caused nonsense-mediated decay. Neither mutation was seen at baseline in the exome sequencing reads, by Sanger sequencing, or by targeted amplicon resequencing. These data suggest that the tumors resistant to anti–PD-1 are a relatively homogenous population derived directly from the baseline tumor and that acquisition of the JAK mutations was an early founder event before clonal selection and relapse despite the fact that the mutation was not detected in pretreatment tumor tissue.
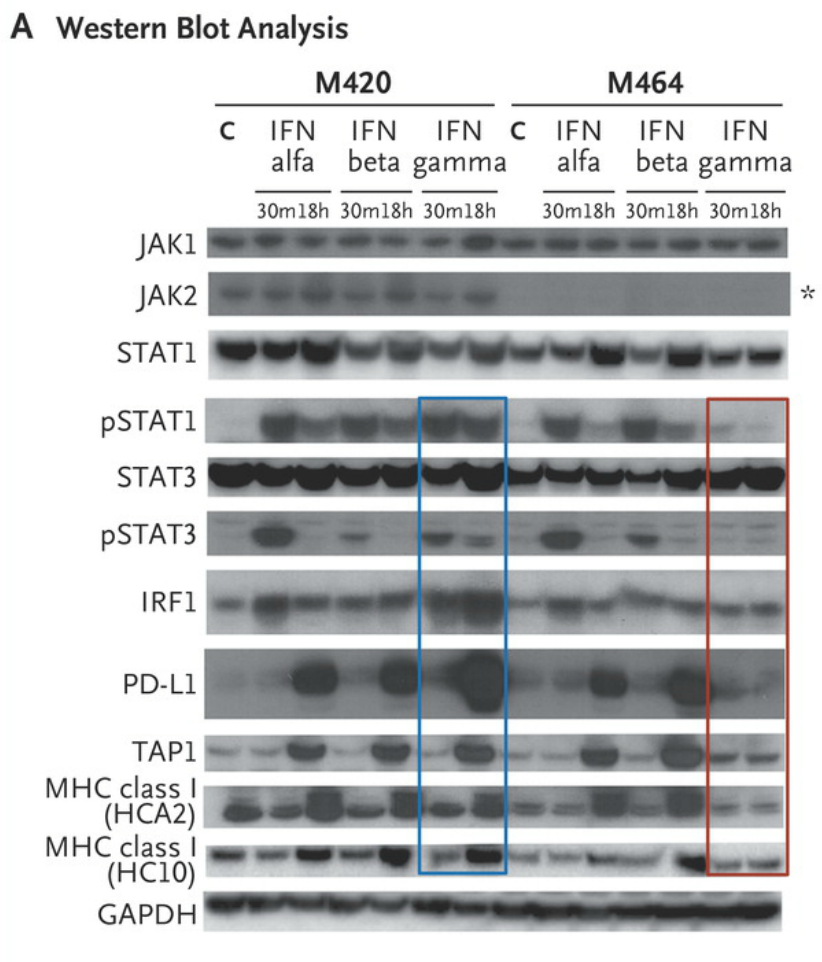
Western blot analysis of lysates from cell lines M420 (Patient 2, baseline) and M464 (Patient 2, relapse) shows Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling events and downstream target induction after either 30 minutes (m) or 18 hours (h) of exposure to interferon (IFN) alfa, beta, or gamma (C indicates untreated control). Janus kinase 2 (JAK2) protein expression was absent in the relapse cell line (asterisk), and M464 failed to phosphorylate intermediate signaling components STAT1 and STAT3 or to up-regulate interferon-response targets TAP1, PD-L1, and major histocompatibility complex (MHC) class I after treatment specifically with interferon gamma (red box), as compared with intact signaling in M420 (blue box). There was no change in response to interferon alfa or beta. As shown in Panel B, a lack of response to interferon gamma exposure was also seen in surface staining for PD-L1 and MHC class I by flow cytometry. Each point represents an independent experiment, T bars represent standard deviations, and Pvaluesare for a two-way analysis of variance with Dunnett’s correction. MFI denotes mean fluorescent intensity, and NS not significant. http://www.nejm.org/doi/full/10.1056/NEJMoa1604958#t=article
The JAK2 mutation was the only homozygous mutation of 76 new nonsynonymous mutations in Patient 2, and the JAK1 mutation was 1 of only 3 homozygous mutations among 53 new mutations in Patient 1. To become homozygous, both JAK mutations were acquired in the context of a copy-number–neutral nondisjunction event, resulting in loss of the wild-type chromosome and duplication of the mutated allele. This is seen clearly in Patient 1: at relapse, chromosome 1p (containing JAK1) showed a decrease in minor-allele frequencies for germline single-nucleotide polymorphisms relative to baseline, was missing 36 heterozygous baseline mutations (presumably on the lost allele), and contained 20 mutations (presumably on the amplified allele) that became homozygous. A similar loss-of-heterozygosity event occurred for chromosome 9 in Patient 2. Together, these data suggest that the tumors resistant to anti–PD-1 are a relatively homogenous population derived directly from the baseline tumor and that acquisition of the JAK mutations was an early founder event before clonal selection and relapse despite the fact that the mutation was not detected in pretreatment tumor tissue.
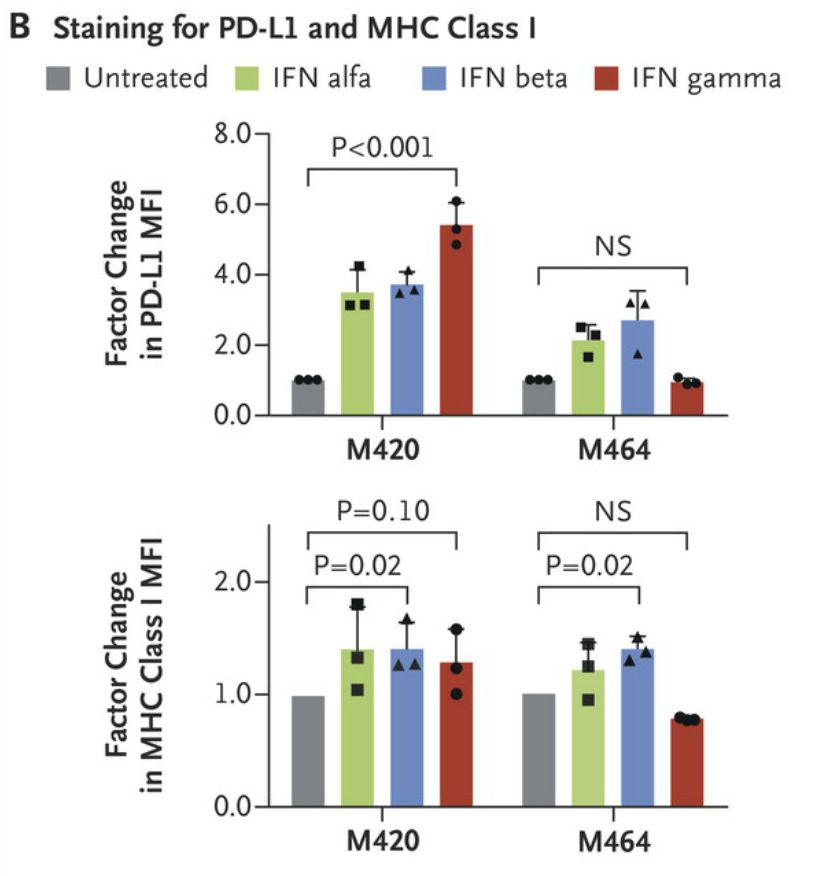
Cell lines M420 (Patient 2, baseline) and M464 (Patient 2, relapse) show a lack of response to interferon gamma exposure was also seen in surface staining for PD-L1 and MHC class I by flow cytometry. Each point represents an independent experiment, T bars represent standard deviations, and Pvaluesare for a two-way analysis of variance with Dunnett’s correction. MFI denotes mean fluorescent intensity, and NS not significant. http://www.nejm.org/doi/full/10.1056/NEJMoa1604958#t=article
Patient 3 – Beta-2-microglobulin mutation
Whole-exome sequencing of the baseline and progressive lesions of tumors from patient 3 showed a 4-bp S14 frame-shift deletion in exon 1 of the beta-2-microglobulin component of MHC class I as 1 of only 24 new relapse-specific mutations and the only such mutation that was homozygous. This finding is in line with the role of beta-2-microglobulin in proper MHC class I folding and transport to the cell surface, and its deficiency has long been recognized as a genetic mechanism of acquired resistance to immunotherapy.
http://www.slideshare.net/drshahinhameed/tests-in-organ-transplantation
Patient 4 – no genetic alternations identified
Researchers could not find defined genetic alterations in Patient 4 that had clear potential to result in acquired resistance to T cells, but cancer cells in the baseline and relapse biopsy samples did not express PD-L1 despite proximity to T cells and PD-L1–expressing stroma. These findings suggest possible non-genetic mechanisms of altered expression of interferon-inducible genes.
Conclusion
The inactivation of JAK1 or JAK2, as noted in two of the patients, may be particularly advantageous to cancer cells in the context of anti–PD-1 therapy as compared with other immunotherapies. Melanomas undergo interferon-induced adaptive expression of PD-L1, which allows the cancer to inactivate adjacent CD8 T cells:
PD-L1 expression on melanoma is largely an adaptive feedback response by melanoma cells to immune attack by TILs. This response limits the effectiveness of immune responses in eradicating melanoma. The actual mechanism of up-regulation of PD-L1 by IFN-γ has not been specifically defined. Additional interest in the expression of PD-L1 on melanoma was generated by the observation that PD-L1 was up-regulated on melanoma cells that had become resistant to treatment with selective BRAF V600 inhibitors. The main pathway involved in this up-regulation was MAPK signalling through MEK but c-Jun and STAT3 proteins also appeared to be required for the expression of PD-L1. Inhibitors of NF-kB signaling down-regulate PD-L1expression on melanoma cells.
When pembrolizumab blocks the the PD-1/PD-L1 axis, IFN-g induced expression of PD-L1 offers no benefit, therefore, the advantage for cancer cells tilts toward abolishing interferon signaling in order to avoid the detrimental increase in antigen presentation and direct anti-proliferative effects of IFN-g. Although inactivating mutations were identified in JAK1 and JAK2, which are receptor-level signaling bottlenecks, interferon insensitivity through other means — such as epigenetic silencing of interferon-signaling components as previously documented in lung-cancer and prostate-cancer cell lines, or increased expression of negative regulators — might lead to the same end. One case of beta-2-microglobulin inactivation was identified; this corroborates a previously described mechanism of acquired resistance to cancer immunotherapy in humans through loss of this shared component of all human MHC class I molecules that is required for CD8 T-cell recognition.
The nearly identical mechanism of acquisition, functional consequence, and evidence of clonal selection for JAK1 or JAK2 mutations in two independent cases with a similar clinical course of acquired resistance suggests that resistance to interferon gamma contributes to immune resistance and escape. This genetic alteration of immune resistance joins the previously described loss of B2M in decreasing immune-cell recognition of cancer cells, leading to acquired resistance to cancer immunotherapy.
Many patients do not respond to treatment with PD-1 inhibition. Insights into the mechanisms of acquired resistance in patients that were sensitive to the therapy, initially, could shed light on the reasons that some do not respond, at all, to PD-1 checkpoint inhibition therapy.
I think testing samples from patients who have failed will allow follow-on therapies that overcome these mechanisms. Ideally, blocking them in the context of checkpoint inhibition and giving therapy that will shed neo-antigens will provoke a significant immune response.
Excellent summary and discussion – thank you for doing this. The key question in the clinic is how we overcome various mechanisms of resistance. Neither JAK1/2 loss of ß2M loss provide much insight into what combinatorial immunotherapy or targeted therapy-immunotherapy strategy might be work. Have you given any thought to how we could move forward theoretically with this precision diagnosis of immunotherapy failure?