

Blood vessel formation (angiogenesis) is an important pathologic process in solid tumors. The liberation of vast amounts of vasoendothelial growth factor (VEGF), which attracts endothelial cells, is responsible for angiogenesis. Since cancer must vascularize in order to grow and metastasize, therapeutic strategies that focus on disrupting the functioning of endothelial cells make great sense for two reasons – (1) endothelial cells are readily accessible, as opposed to many areas of the tumor, itself; and (2) they are also genetically stable, therefore less likely to develop mutations and resistance compared to cancer cells, which have highly unstable genomes.
What is Endoglin and what is its role?
Endoglin is a superfamily of homodimeric transmembrane proteins which are major glycoproteins of the vascular endothelium. This protein is a component of the transforming growth factor beta receptor complex (CD105) and it binds to the beta1 and beta3 peptides with high affinity; it has antagonistic effects to TGF-β. Mutations in this gene cause hereditary hemorrhagic telangiectasia, also known as Osler-Rendu-Weber syndrome 1 (Hereditary Hemorrhagic Telangiectasia, or HHT), an autosomal dominant multi-systemic vascular dysplasia. This gene may also be involved in preeclampsia and several types of cancer.
The role of endoglin may be to conserve stem cell potential, versus differentiation of precursor cells, via its modulation of TGF-β and bone morphogenetic protein (BMP). Endoglin is expressed on mesenchymal cells and is active in embryogenesis during the endocardial-mesenchymal transformation that leads to heart septation and valve formation. Endoglin-null embryos die at mid-gestation due to defects in vessel and heart development, indicating that endoglin is essential for both angiogenesis and heart development (Figure 1). Endoglin is also present on fibroblasts at the edge of a wound and on perivascular stromal cells involved in vascular remodeling. Additionally, endoglin is found on vascular smooth muscle cells, particularly after arterial injury, and could mediate the effects of TGF-β on their migration and tissue repair.

Figure 1 – top left panel, shows blood vessels expressed in mice with endoglin; right panel shows absence of blood vessels in endoglin knock out mice. file:///C:/Users/jvgul/Downloads/TRACON%20Investor%20Deck%20(April%202016),%20Final.pdf
Why was Endoglin blockade considered a viable strategy for cancer treatment?
Providing a hint that blocking endoglin may be a viable strategy for cancer therapy is the finding that patients with HHT who develop cancer fare better than those without HHT (Figure 1). In patients who were diagnosed with breast, colorectal, lung, or prostate cancer between 2000 and 2007, those with a concomitant diagnosis of HHT had a significantly increased survival (Fig. 1, P <0.0001). For individual cancers, breast cancer showed the strongest association between HHT diagnosis and survival.
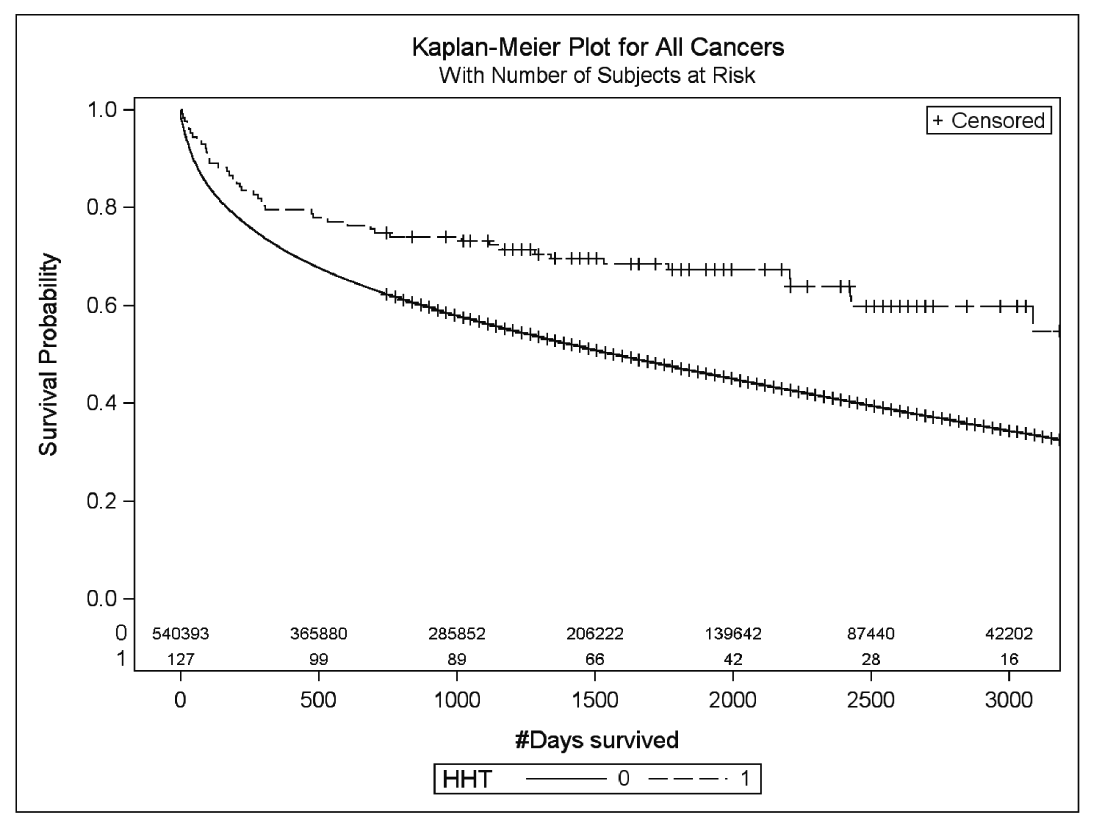
Figure 2 – Kaplan-Meier survival curves for a composite of the four cancers (breast, prostate, lung, and colorectal) and a diagnosis of HHT. Diagnosis of normal non-HHT or HHT is indicated by 0
and 1, respectively. Log-rank P value is indicated in the figure. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3947104/
How Does Endoglin Work?
This superfamily receptor complex also includes activin-receptor-like kinases (ALK) and bone morphogenic protein receptor (BMPR) as well – Figure 3. In hypoxic conditions endoglins are over expressed through induction of hypoxia inducible factor 1-α. In the absence of endoglin, activation of TGF-β receptors on quiescent endothelium leads to SMAD 2 and 3 phosphorylation which inhibit endothelial cell growth and proliferation. SMAD proteins transduce extracellular signals from TGF-β ligands to the nucleus where they activate downstream gene transcription. However, the binding of bone morphogenetic protein (BMP) to TGF-β receptor complex on proliferating endothelium results in phosphorylation of SMADs 1, 5 and 8, which activates cell growth and release of growth inhibitory proteins SMAD 2 and 3. High endoglin content detected on vessels are correlated with poor prognosis in many solid tumors such as breast, lung, prostate and colon.
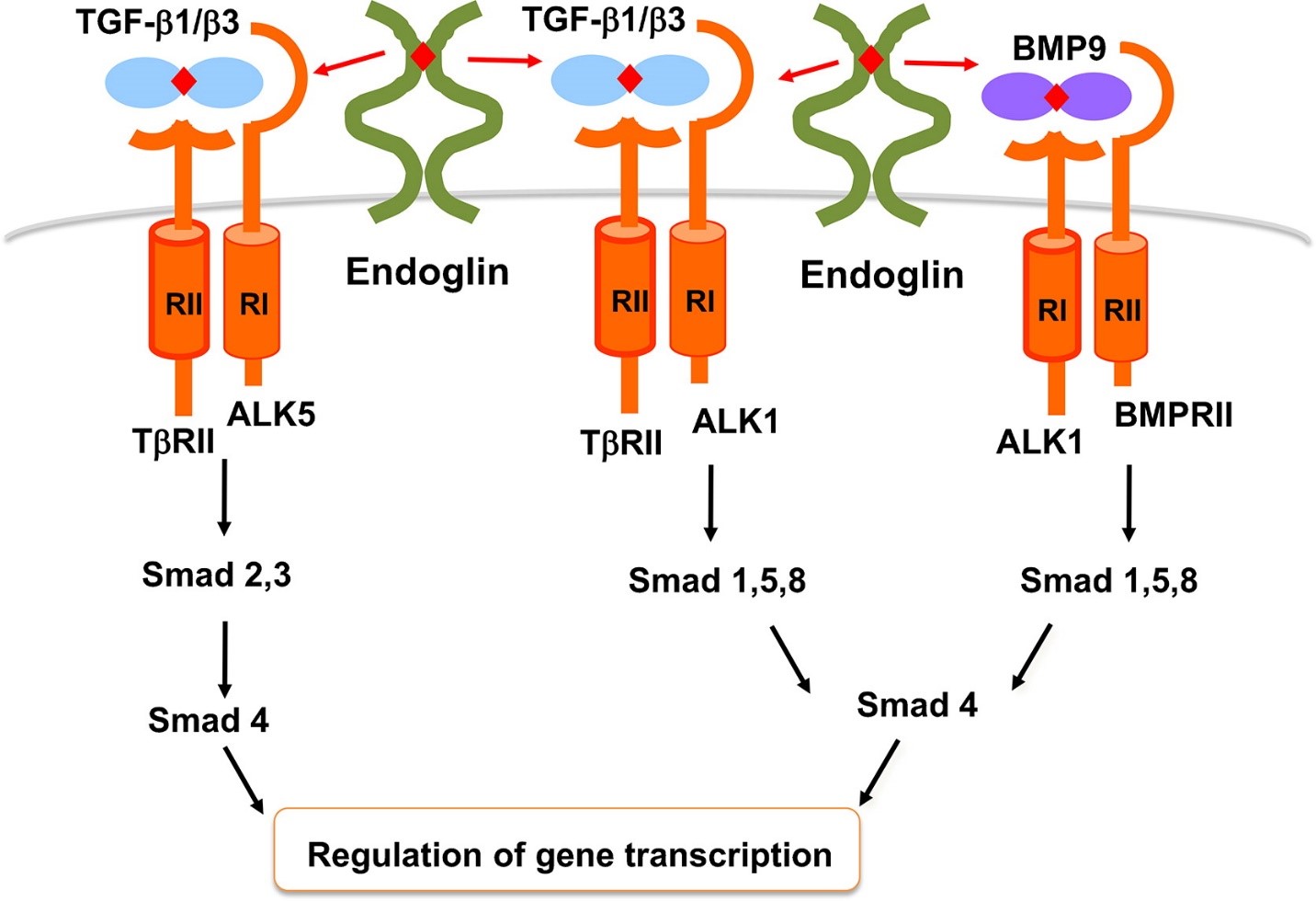
Figure 3: TGF-β and BMP9 signaling pathways in endothelial cells. In endothelial cells, TGF-β1 and –β3 bind to TβRII which then recruits and phosphorylates ALK5 or ALK1. These two type I receptors signal via Smad 2,3 and Smad 1,5,8 respectively. Smad4 is responsible for transfer of Smad complexes to the nucleus where they regulate gene transcription. BMP9 binds to ALK1 and BMPRII and signals via Smad 1,5,8 pathway. Endoglin can modulate all of these pathways in a cell and context-dependent manner. http://www.placentajournal.org/article/S0143-4004(13)00793-5/fulltext
The precise function of endoglin in TGF‐β/ALK signalling has remained unclear. Endoglin is required for efficient TGF‐β/ALK1 signaling, which indirectly inhibits TGF‐β/ALK5 signaling. Endothelial cells lacking endoglin do not grow because TGF‐β/ALK1 signaling is reduced and TGF‐β/ALK5 signaling is increased. Surviving cells adapt to this imbalance by downregulating ALK5 expression in order to proliferate. The ability of endoglin to promote ALK1 signaling also explains why ectopic endoglin expression in endothelial cells promotes proliferation and blocks TGF‐β‐induced growth arrest by indirectly reducing TGF‐β/ALK5 signaling. Endoglin has a pivotal role in the balance of ALK1 and ALK5 signaling to regulate endothelial cell proliferation. Blocking endoglins with monoclonal antibodies can arrest angiogenesis.
Anti-Endoglin Monoclonal Antibody Therapy Rationale
Treatment of patients with anti-VEGF antibodies (and tyrosine kinase inhibitors) has proven to be very effective. Bevacizumab, which targets VEGF, is a very successful product, called Avastin (by Genentech). that is approved for use in colorectal, kidney, lung, cancer and glioblastoma. Ramucirumab (Cyramza, by Lilly) binds to the VEGF Receptor-2, and is approved for the treatment of stomach cancer. Sorafenib (Nexavar by Bayer) is a VEGFR 1,2,3 receptor tyrosine kinase (RTK) inhibitor approved for the treatment of renal cell cancer, hepatocellular carcinoma, and thyroid cancer; pazopanib (Votrient by Novartis) is a VEGF1,2,3 RTK inhibitor approved for soft tissue sarcoma and renal cell carcinoma. Resistance to anti-VEGF therapy is developed through emergence of escape pathways, rather than mutations to the VEGF receptor or its ligands. It has been shown that endoglins are upregulated when VEGF inhibition occurs during hypoxia. Endoglin monoclonal antibodies function by targeting the dominant escape pathway which causes anti-VEGF resistance, hence combining anti-endoglin antibody therapy with anti-VEGF treatment is an attractive approach.
TRC105 developed by Tracon Pharmaceuticals, is a novel human chimeric IgG1 monoclonal antibody that binds to the orphan domain of the endoglin (Figure 4). This monoclonal antibody prevents BMP binding to the BMPR thereby preventing SMAD 1, 5 and 8 phosphorylation, then recruits SMAD 2 and 3 for growth inhibition. In addition to blocking, endoglin, TRC105 also induces antibody dependent cell mediated cytotoxicity (ADCC), which results in endothelial cell death, another mechanism by which it arrests angiogenesis.
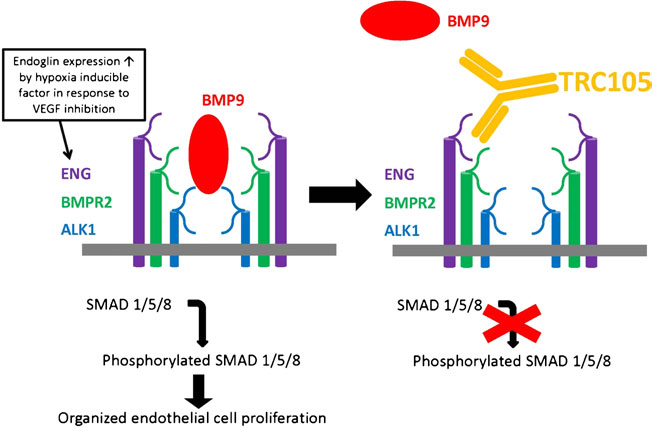
Figure 4: TGF-β and BMP9 signaling pathways in endothelial cells. In endothelial cells, TGF-β1 and –β3 bind to TβRII which then recruits and phosphorylates ALK5 or ALK1. These two type I receptors signal via Smad 2,3 and Smad 1,5,8 respectively. Smad4 is responsible for transfer of Smad complexes to the nucleus where they regulate gene transcription. BMP9 binds to ALK1 and BMPRII and signals via Smad 1,5,8 pathway. Endoglin can modulate all of these pathways in a cell and context-dependent manner. http://www.placentajournal.org/article/S0143-4004(13)00793-5/fulltext
Clinical Results with TRC105
Tracon Pharmaceuticals is currently conducting several phase 1/2 trials for breast cancer, renal cell carcinoma, glioblastoma, hepatocellular carcinoma and soft tissue sarcoma and a phase 1 trial for lung carcinoma with TRC105 and different VEGF inhibitors. In a phase 1B study of TRC105 combined with bevacizumab in patients with advanced solid malignancies, telangiectasia (small dilated blood vessels visible on the skin or mucous membranes), an “on target” manifestation of endoglin modulation, was dose-dependent. Epistaxis (nose bleeds), another on-target manifestation of endoglin modulation, was the most common adverse event in a phase 1b study of TRC105 and pazopanib in patients with sarcoma.
The most striking results were obtained from the combined TRC105 therapy in patients who were refractory to prior therapy with VEGF inhibitors. Around 25% of patients who were resistant to bevacizumab and tyrosine kinase inhibitors responded to TRC 105 therapy plus bevacizumab. CT scans demonstrated decreased tumor density and the patients exhibited longer progression free intervals with the combined treatment versus prior VEGF inhibitor therapy (Figures 5 and 6).
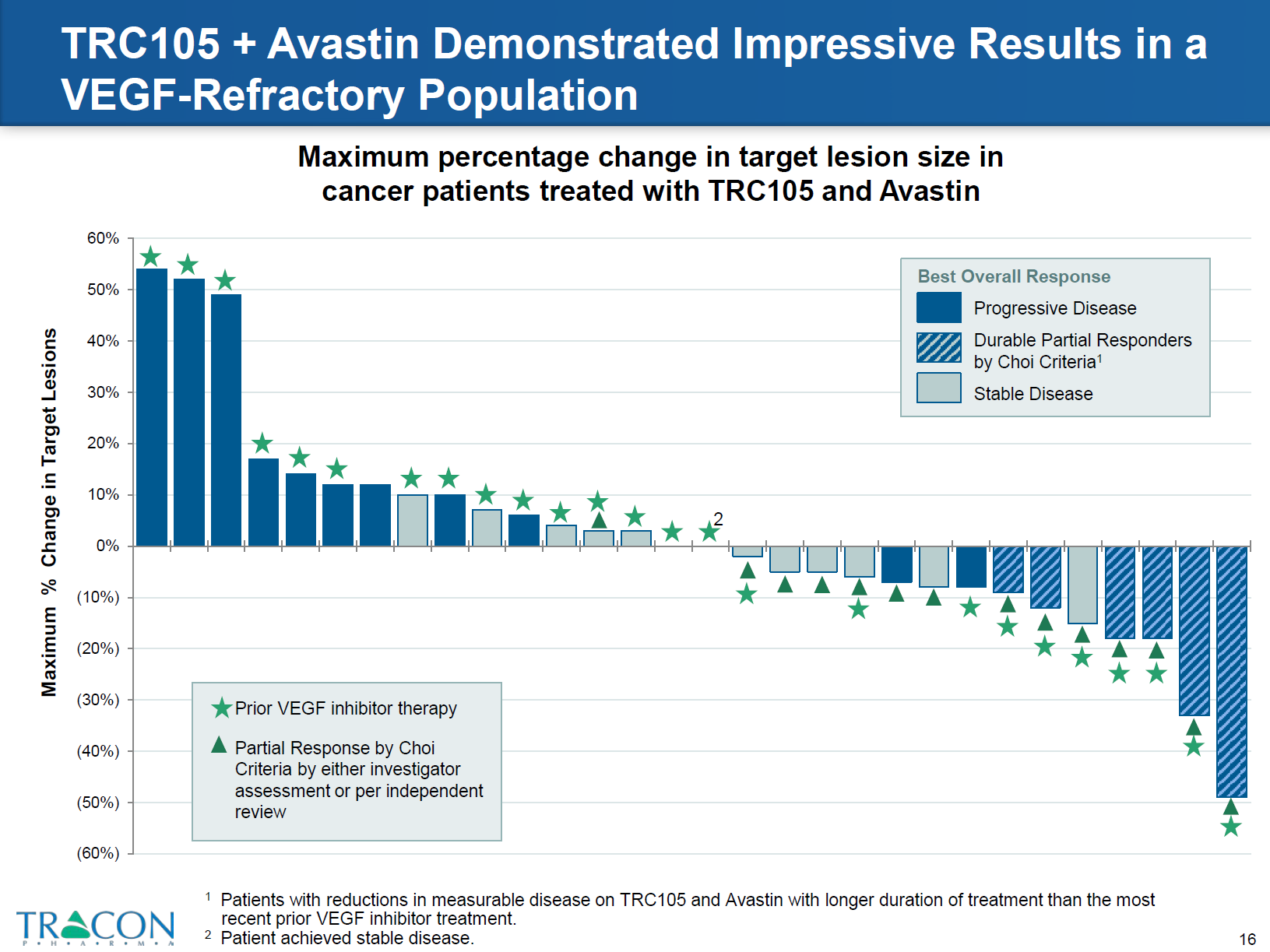
Figure 5: file:///C:/Users/jvgul/Downloads/TRACON%20Investor%20Deck%20(April%202016),%20Final.pdf
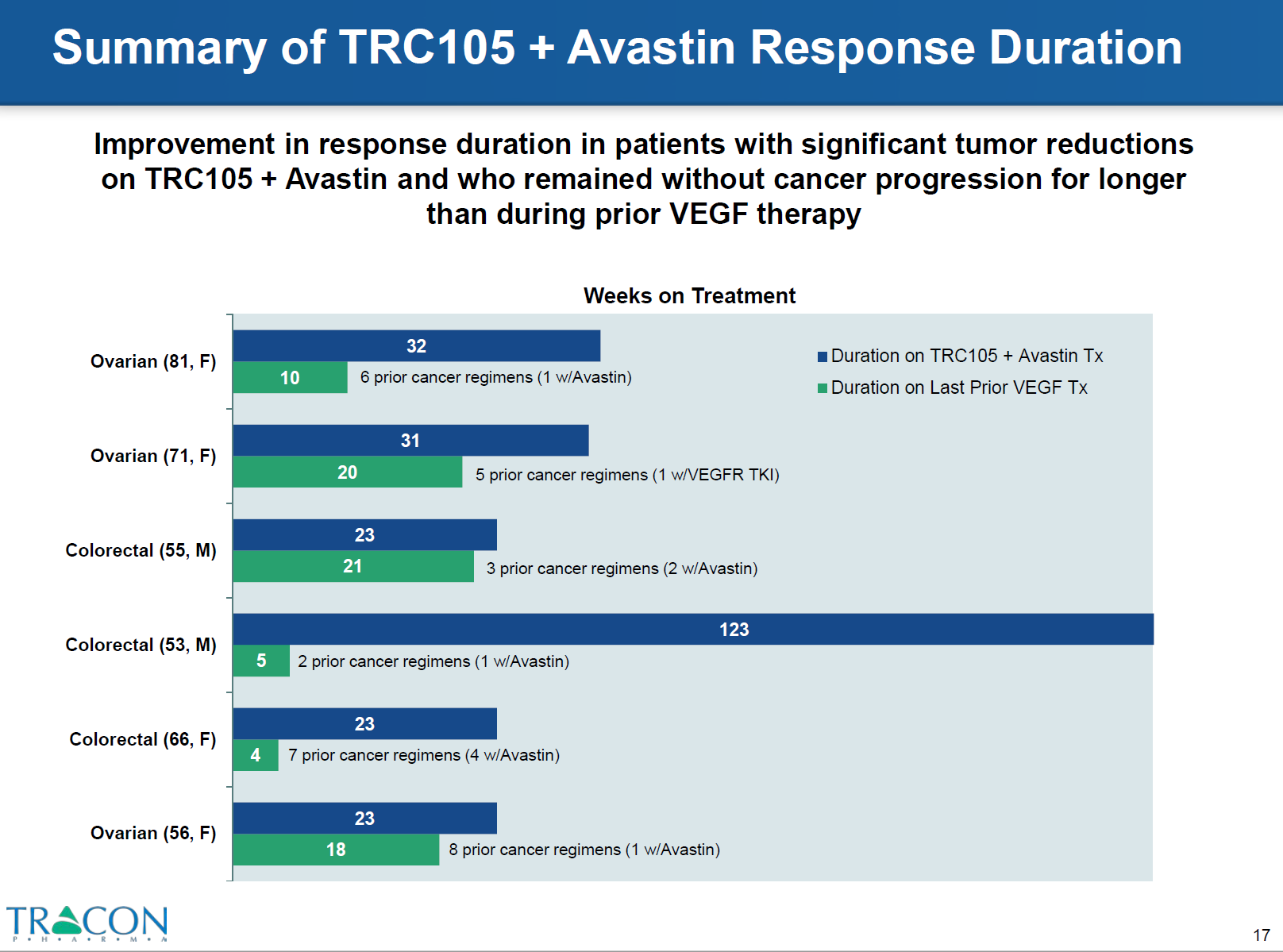
Figure 6: file:///C:/Users/jvgul/Downloads/TRACON%20Investor%20Deck%20(April%202016),%20Final.pdf