Cancer results from accumulated mutations in the cancer cell’s genome. These mutations can occur spontaneously in any cell throughout an individual’s lifetime, often increasing with age or exposure to carcinogenic or mutagenic compounds. These are called somatic mutations that do not exist in every cell, and cannot be passed along from one generation to the next. However, in approximately 5 to 10% of all cancer cases, mutations are passed along through the germ line and can predispose an individual to various types of cancers. While these individuals with inherited mutations are more likely to get cancer at some point in their lifetime, it is not a guarantee. Cancer inheritance patterns can be autosomal dominant or autosomal recessive with the cancer phenotype typically being recessive –requiring two mutant alleles to show the cancer phenotype. Inheriting one mutant allele does not necessarily transform the cell, but it does put the individual one step closer to malignant transformation. Typically, multiple somatic genetic events (mutation, promoter silencing, loss of heterozygosity, etc.) must ensue before and an inherited mutation can lead to cancer.
Gatekeepers versus Caretakers
Many inherited cancer syndromes are due to mutant tumor suppressor genes. Many tumor suppressor genes are responsible for controlling the cell cycle by either repairing damage, or initiating apoptosis. Tumor suppressor genes can fall into one of two categories: gatekeeper genes and caretaker genes (Figure 1). Gatekeeper genes allow or disallow a cell to progress through the cycles of growth and proliferation. Caretaker genes are responsible for genome maintenance and can repair damage to the cell’s DNA. Both are critical mechanisms to prevent the proliferation of cells with a highly mutable genome, which can potentially lead to cancer cell transformation. Tumor suppressor gene mutations are easily transferred through the germ-line since their presence does not have a major impact on embryonic development. [The opposite occurs with oncogenes, which are autosomal dominant and drive the cell cycle – pregnancies rarely proceed to full term.]
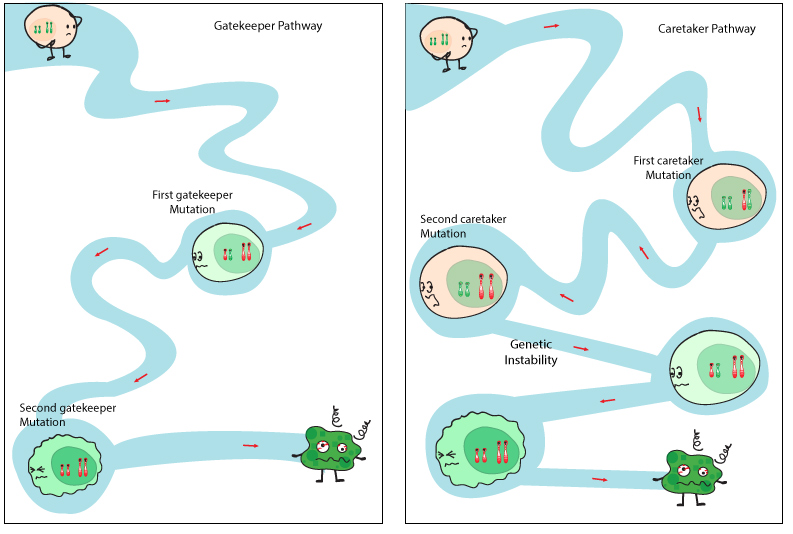
Mutations in both caretaker and gatekeeper pathways lead to tumour initiation through a two-hit model. Dysfunctional caretaker genes can lead to rapid acquisition of mutations in several gatekeeper genes. Released under the Creative Commons Attribution-ShareAlike 4.0 International license (CC BY-SA 4.0). http://www.cubocube.com/dashboard.php?a=347&b=428&c=1
Tumor suppressor gene mutations in the germ line tend to “lay low” until something goes wrong and their lack of activity perpetuates or magnifies the subsequent genetic insult. Inheritance of only one mutated allele allows the wild type allele to keep the other in check. This post will focus on three main tumor suppressor genes responsible for many inherited cancer syndromes: PTEN, BRCA 1 & 2, and TP53.
PTEN
PTEN is a gatekeeper tumor suppressor gene responsible for the control of the PI3K pathway – a downstream effector of Ras – and is linked with Cowden syndrome, which is an inherited disorder that results in many noncancerous tumors throughout the body. Individuals suffering from Cowden’s syndrome are at an increased risk for developing various forms of cancer including breast and thyroid.
The PI3K pathway is critical to cell growth and proliferation and it also inhibits apoptosis, which makes it an ideal target for cancer cells. PI3K is responsible for attaching a third phosphate onto phosphatidylinositol (PIP2) that is bound to the plasma membrane. By adding this third phosphate, PIP2 is converted into PIP3. PIP3 is in ideal formation to dock to a PH domain for several types of cytoplasmic proteins such as Akt/PKB. The attachment of Akt/PKB to PIP3 via its PH domain allows for the activation of Akt/PKB. Functioning as a kinase, Akt/PKB proceeds to phosphorylate protein substrates with multiple effects on the cell, including proliferation, inhibition of apoptosis, motility, and angiogenesis (Figure 2). PTEN’s role involves the removal of one of the phosphates on PIP3 converting it back to PIP2. Without the third phosphate, PIP2 cannot effectively bind to the PH domain of Akt/PKB subsequently halting the entire proliferative process. Inactivating PTEN via mutation or methylation of CpG sequences deregulates the PI3K pathway constantly allowing it to activate downstream effectors.
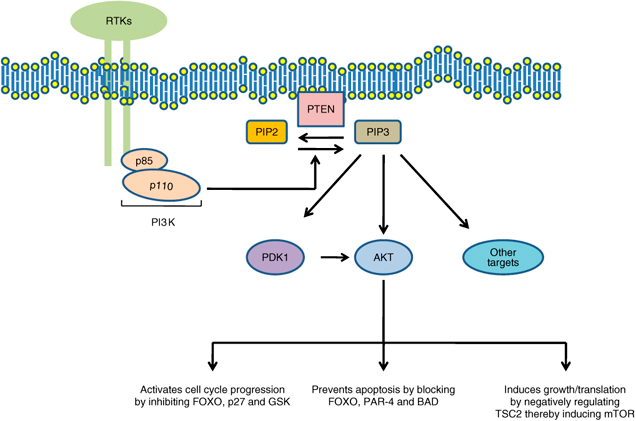
PTEN’s role in the PI3K pathway. Without PTEN converting PIP3 back to PIP3, a number of downstream effectors that induce proliferation, growth and anti-apoptotic proteins are activated
The role of PTEN signaling perturbations in cancer and in targeted therapy M Keniry and R Parsons http://www.nature.com/onc/journal/v27/n41/fig_tab/onc2008248f1.html
BRCA1 & BRCA2
BRCA 1 and BRCA 2 are caretaker tumor suppressor genes. As mentioned above, caretaker genes maintain genomic integrity by fixing errors – predominantly associated with dsDNA breaks at replication forks. The BRCA family of genes works in concert with other proteins to form large complexes that initiate homology-directed repair (HDR) to fix dsDNA breaks. With a mutated BRCA 1 or 2, the cell cannot repair the stand breaks, which leads to an accumulation of mutations that drive proliferation and metastases. Much is still unknown about BRCA 1 and 2, especially why loss of function results primarily in breast and ovarian cancers. BRCA1 mutation carriers have a lifetime risk of breast cancer that is greater than 80%. The ultimate lifetime breast cancer risk forBRCA2 mutation carriers approaches that of BRCA1 carriers; however, a later age of disease onset has been documented for BRCA2 mutation carriers. In addition to breast cancer, women with BRCA1 mutations have an increased risk of ovarian cancer and, to a much lesser extent, males have an increased risk of prostatic cancer. BRCA2 mutation carriers are at increased risk of breast cancer in males and females, and of ovarian, prostatic, pancreatic, gall bladder, bile duct and stomach cancers and melanoma.
BRACA1 also mediates cell cycle arrest – BRCA1-depleted cells have defective G1/S checkpoint in response to DNA damage. BRCA1 acts as a scaffold protein facilitating phosphorylation of p53 by ATM in response to DNA damage, which led to p53-mediated induction of p21 and induced G1/S arrest.
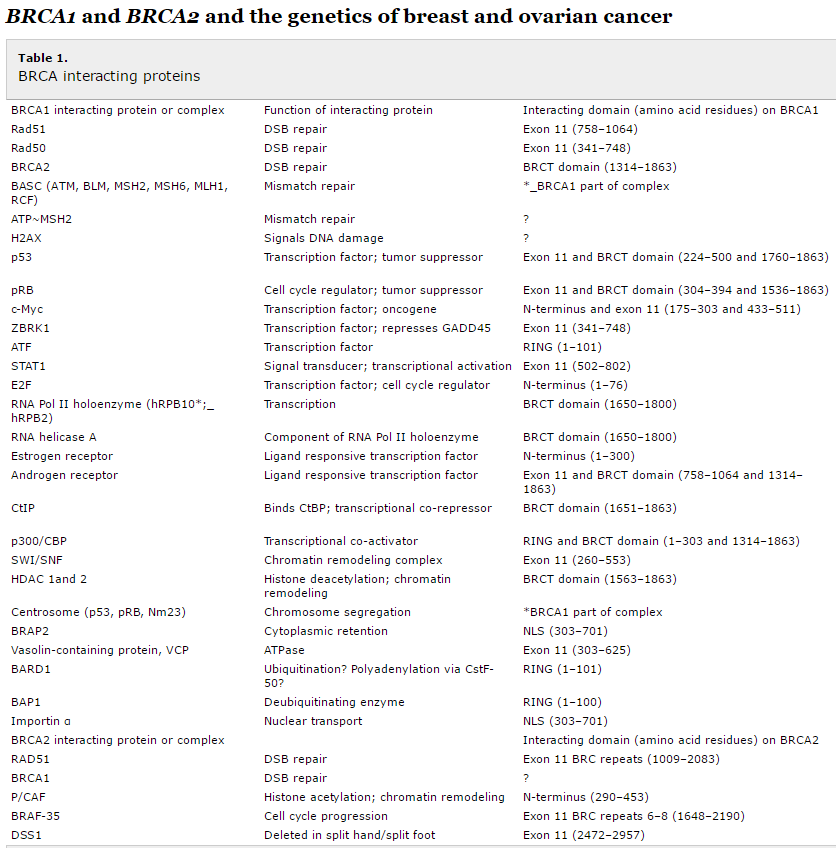
Table 1 lists the proteins with which BRCA1 and BRCA2 interact.
TP53
TP53 is both a caretaker and gatekeeper gene – it regulates the cell cycle by orchestrating a series of events to either pause the cell cycle and initiate repair of mistakes found in the genome, or abort all repair efforts and destroy the cell before it takes on potentially catastrophic properties.
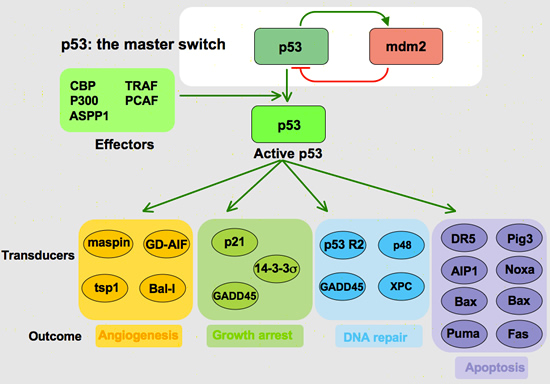
TP53 acts as a transcription factor by inducing proteins to affect cell cycle arrest and apoptosis (Figure 3). Most mutations of TP53 occur at its DNA binding site, which prohibits it from completing its job as a transcription factor. One of the proteins initiated by TP53 is p21 – a Cyclin Dependent Kinase Inhibitor (CDK). CDKs are responsible for allowing a cell to progress through the cell cycle. If the CDK is blocked, then the cell cycle grinds to a halt and prevents any further proliferation or replication of damaged DNA. Other proteins governed by TP53 are Siah-1, which is responsible for degrading β – catenin — a protein that induces the expression of cyclin D1, another critical component to cell cycle progression.
TP53 also determines when a cell is beyond repair and initiates apoptosis. Again acting as a transcription factor, TP53 induces the expression of a variety of intrinsic pro-apoptotic proteins such as Bax and PUMA. These proteins open cyctochrome C channels in the mitochondria. Once cytochrome C is in the cytoplasm, it activates caspases that cleave “death substrates.” The degradation of these “death substrates” initiates a cascade of cellular alterations associated with apoptosis. Another intrinsic factor influenced by TP53 is the FOXO3 transcription factor; yet another pro-apoptotic protein.
TP53 also induces the expression of Insulin-like Growth Factor Binding Protein (IGFBP-3), which binds to Insulin-like Growth Factor (IGF) and prevents it from binding with its receptor, IGF-R1. This binding inhibition blocks “survival signals” to the cell, which further allows the proverbial scale to tip in the favor of apoptosis.
TP53 does not only govern intrinsic apoptotic factors, but extrinsic as well by inducing the expression of the gene that codes for the FAS receptor. When FAS ligand binds with its receptor on the surface of a cell, a cascade of caspases are activated to yet again cleave “death substrates”.
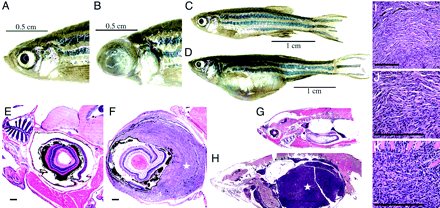
As one can see, an alteration in the TP53 gene can result in severe consequences. Under normal conditions, a cell that has accumulated dangerous mutations will self-destruct before it has an opportunity to transform. Individuals with an inherited mutation in this critical tumor suppressor gene accumulate potentially transformative mutations in their cells because repair is hampered. Zebrafish with mutant P53 has a predisposition for developing tumors of the eye and abdomen (Figure 4). Inheritance of a mutant TP53 in humans results in Li-Fraumeni syndrome, which is a disorder that predisposes individuals to breast cancer, osteosarcoma, leukemia, brain tumors, and other cancers.