

BET bromodomain inhibitors are epigenetic regulators that act by repressing expression of oncogenes, including MYC and BCL-2, that are aberrantly highly expressed in many cancer cells, resulting in inhibited cell proliferation and induction of apoptotic cell death.
What is Epigenetics?
The term epigenetics refers to heritable changes in gene expression (active versus inactive genes) that does not involve changes to the underlying DNA sequence; a change in phenotype without a change in genotype. Epigenetic change is a regular and natural occurrence but can also be influenced by several factors including age, the environment/lifestyle, and disease state. Epigenetic modifications can manifest as commonly as the manner in which cells terminally differentiate to end up as skin cells, liver cells, brain cells, etc. Or, epigenetic change can have more damaging effects that can result in diseases like cancer. At least three systems are currently considered to initiate and sustain epigenetic change:
- DNA methylation,
- Histone modification and
- non-coding RNA (ncRNA)-associated gene silencing
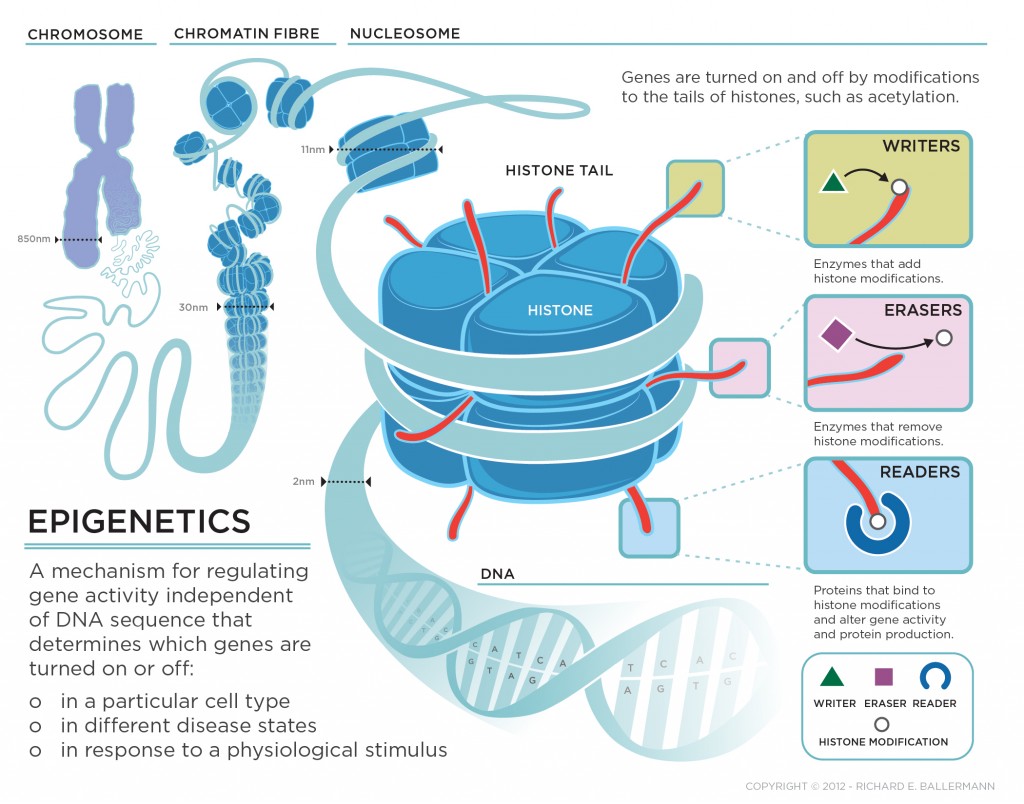
Writers, Readers, Erasers, and Super Enhancers
“Writers” are proteins that add molecules to histones, for example, histone acetylransferases (HAT’s – make chromatin more open, therefore, more transcription-friendly), histone methyltransferases (HMT’s – make chromatin more compact), protein arginine methyltransferases (PAMT’s – block the docking site for readers and other epigenetic effectors). “Erasers” are proteins that remove molecules from histones, for example, histone deacetylases (HDACs), lysine demethylases (KDMs) and phosphatases. Readers are proteins containing bromodomains, chromodomains and Tudor domains that bind to epigenetic marks (acetyl, methyl, phosphate, and arginine groups) on histones and regulate gene expression.
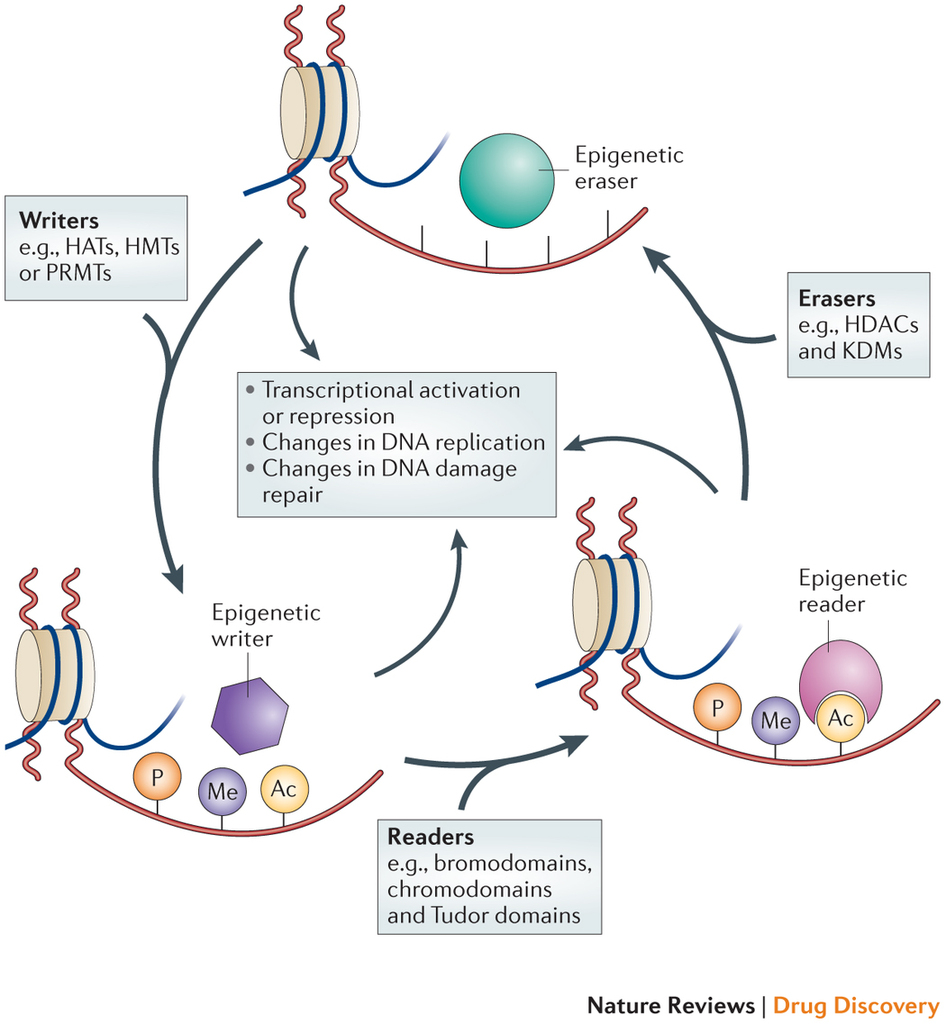
Epigenetic regulation is a dynamic process. Epigenetic writers such as histone acetyltransferases (HATs), histone methyltransferases (HMTs), protein arginine methyltransferases (PRMTs) and kinases lay down epigenetic marks on amino acid residues on histone tails. Epigenetic readers such as proteins containing bromodomains, chromodomains and Tudor domains bind to these epigenetic marks. Epigenetic erasers such as histone deacetylases (HDACs), lysine demethylases (KDMs) and phosphatases catalyse the removal of epigenetic marks. Addition and removal of these post-translational modifications of histone tails leads to the addition and/or removal of other marks in a highly complicated histone code. Together, histone modifications regulate various DNA-dependent processes, including transcription, DNA replication and DNA repair. http://www.nature.com/nrd/journal/v13/n9/fig_tab/nrd4360_F1.html
Super-enhancers are large clusters of transcriptional enhancers that drive expression of genes that define cell identity. Super-enhancers associate with genes that control and define the biology of cells. Interestingly, disease-associated variation is especially enriched in the super-enhancers of disease-relevant cell types. Cancer cells generate super-enhancers at oncogenes and other genes important in tumor pathogenesis.
Cancer progression can be driven by tumor cells acquiring super-enhancers to overexpress certain hallmark oncogenes. BET inhibitors selectively suppress super-enhancer driven gene expression, and have shown selectivity towards tumor cells. BET proteins also play a central role in the transcriptional program of oncogenes driven by gene fusions, translocations and mutations.
Drug resistance is a critical limitation across all therapeutic modalities and there is a broad preclinical dataset demonstrating that the treatment of these resistant cancers with BET inhibitors renders them sensitive to the various therapies. This offers significant clinical potential for the treatment of resistant cancers with BET inhibitors.
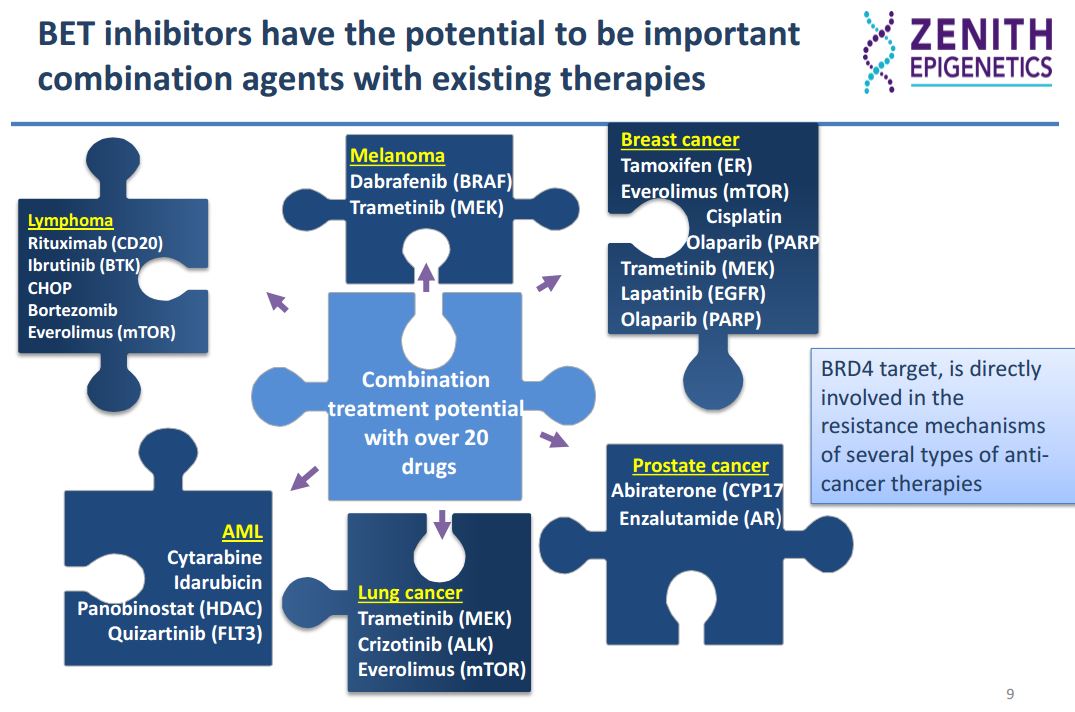
BRD4
BRD4 (bromodomain-containing protein 4) is a chromatin reader protein that recognizes and binds acetylated lysine residues in histones and plays a key role in transmission of epigenetic memory across cell divisions and transcription regulation. It remains associated with acetylated chromatin throughout the entire cell cycle and provides epigenetic memory for postmitotic G1 gene transcription by preserving acetylated chromatin status and maintaining high-order chromatin structure. During interphase, BRD4 plays a key role in regulating the transcription of signal-inducible genes by associating with the P-TEFb complex and recruiting it to promoters: BRD4 is required to form the transcriptionally active P-TEFb complex by displacing negative regulators such as HEXIM1 and 7SKsnRNA complex from P-TEFb, thereby transforming it into an active form that can then phosphorylate the C-terminal domain (CTD) of RNA polymerase II. It promotes phosphorylation of ‘Ser-2’ of the C-terminal domain (CTD) of RNA polymerase II. It is released from chromatin upon deacetylation of histones that can be triggered by different signals such as activation of the JNK pathway or nocodazole treatment.
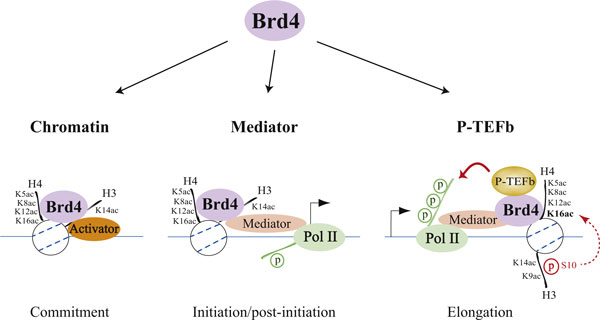
Acetylation of histone H3 and H4 lysine residues modulates Brd4 association with chromatin and the recruitment of Mediator and P-TEFbThree steps for bromodomain-containing protein 4 (Brd4)-regulated chromatin targeting and transcriptional regulation are highlighted. The first step (left) represents a commitment to target gene transcription illustrated by cooperative binding between Brd4 and a transcriptional activator with acetylated chromatin through Brd4-activator interaction, activator-DNA contact, and Brd4 association, via its tandem bromodomains, with acetylated lysine 5 (K5ac), acetylated lysine 8 (K8ac), acetylated lysine 12 (K12ac), and acetylated lysine 16 (K16ac) of histone H4, and/or acetylated lysine 14 (K14ac) of histone H3. The second step (center) is Brd4-mediated recruitment of the initiation cofactor Mediator to the promoter region, which often leads to phosphorylation of the RNA polymerase II (Pol II) carboxyl-terminal domain (CTD) at Ser5 during initiation and post-initiation events. The third step (right) is Brd4-facilitated recruitment of the elongation cofactor P-TEFb (positive transcription elongation factor b) to paused Pol II that results in Ser2 phosphorylation of the CTD, thereby allowing Pol II to resume elongation. The inducible recruitment of Brd4 to an acetylated nucleosome located downstream of the transcription start site (indicated by an arrow) appears to depend on crosstalk between acetylated lysine 9 (K9ac) and phosphorylated serine 10 (S10) of H3 with H4K16ac. http://f1000.com/prime/reports/b/1/98/fig-002
BRD4 Inhibition in Prostate Cancer
Amplification of the MYC locus is frequent in prostate cancer (30%) and is associated with disease progression and Gleason score. c-Myc protein levels are also elevated in prostate tumors compared to normal prostate epithelium, with a stepwise increase from low-grade to high-grade prostatic intraepithelial neoplasia (PIN). MYC overexpression is sufficient to transform benign human prostate epithelium in vitro and MYC transgenic mice develop PIN , suggesting that MYC plays a role in prostate cancer initiation. MYC is a promising target in prostate cancer as demonstrated in the preclinical models of prostate cancer with antisense nucleotides.
The highly specific BET inhibitor, I-BET762, was evaluated on MYC expression in prostate cancer models. I-BET762 potently reduced MYC expression in prostate cancer cell lines and a patient-derived tumor model with subsequent inhibition of cell growth and reduction of tumor burden in vivo.
In the AR (androgen-receptor) positive VCAP prostate cancer cell line, ZEN-3694 (a small molecule that selectively binds to both bromodomains of the BET proteins, inhibiting the interaction of acetylated histone peptide with IC50 values in low nM range) inhibits proliferation synergistically with the AR antagonist enzalutamide.
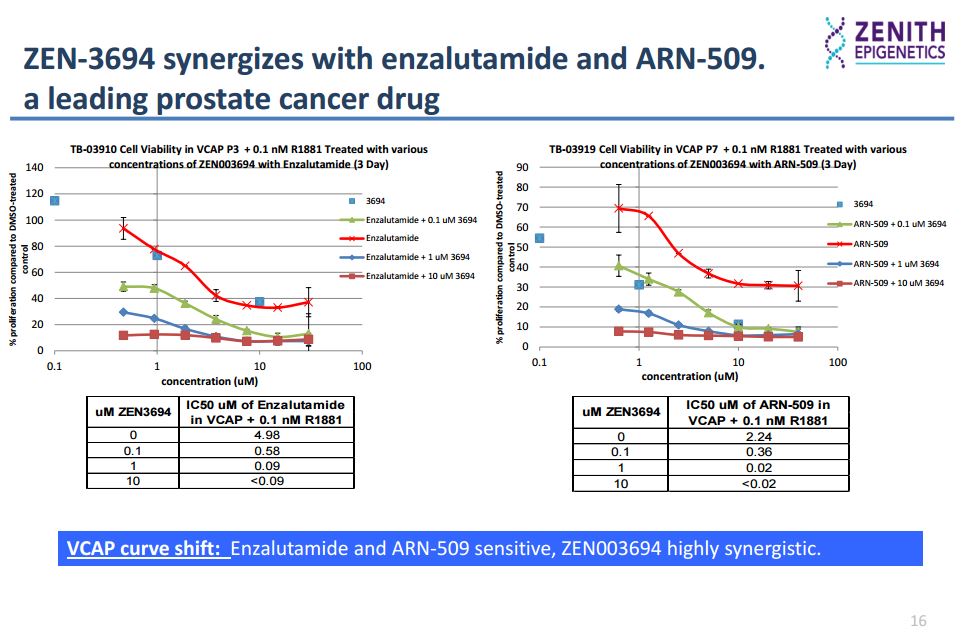
In an in vitro enzalutamide resistance model characterized by the up-regulation of the glucocorticoid receptor (GR), GR expression was inhibited by ZEN-3694 in a dose-dependent manner. Sensitivity to ZEN-3694 was unaltered, suggesting that it could be a valid therapeutic approach in patients developing resistance to AR antagonists through GR induction.
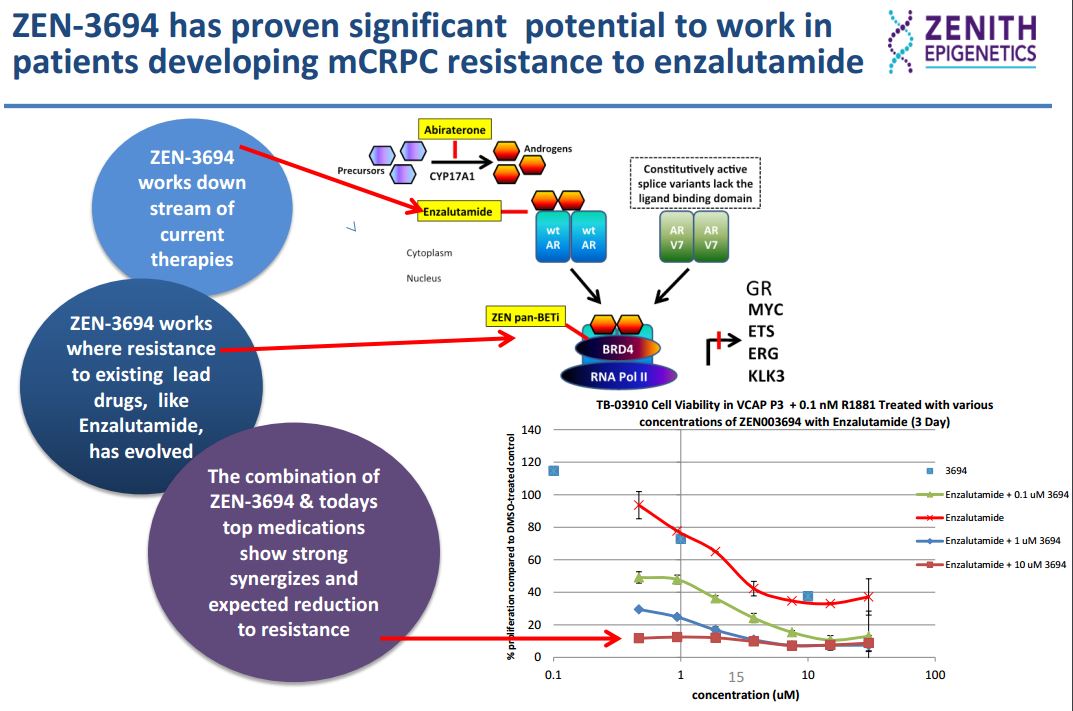
Clinical trials of ZEN-3694 in patients with metastatic resistant prostate cancer will commence in 2016.