

Adult acute myeloid leukemia (AML) is a cancer of the blood and bone marrow. This type of cancer usually gets worse quickly if it is not treated.
Types of Leukemia
An estimated 54,270 new cases of leukemia are expected in 2015. Leukemia is a cancer of the bone marrow and blood and is classified into four main groups according to cell type and rate of growth:
- acute lymphocytic (ALL),
- chronic lymphocytic (CLL),
- acute myeloid (AML), and
- chronic myeloid (CML).
The majority (91%) of leukemia cases are diagnosed in adults 20 years of age and older. Among adults, the most common types are CLL (36%) and AML (32%). In contrast, ALL is most common before age 20, accounting for 76% of cases. Overall leukemia incidence rates have been slowly increasing over the past few decades; from 2007 to 2011, rates increased by 1.6% per year in males and 0.6% per year in females. The current 5-year relative survival of 25% for patients diagnosed with AML.
Acute Myeloid Leukemia
In AML, the myeloid stem cells usually become a type of immature white blood cell called myeloblasts (or myeloid blasts). The myeloblasts in AML are abnormal and do not become healthy white blood cells. Sometimes in AML, too many stem cells become abnormal red blood cells or platelets. These abnormal white blood cells, red blood cells, or platelets are also called leukemia cells or blasts. Leukemia cells can build up in the bone marrow and blood so there is less room for healthy white blood cells, red blood cells, and platelets. When this happens, infection, anemia, or easy bleeding may occur. The leukemia cells can spread outside the blood to other parts of the body, including the central nervous system (brain and spinal cord), skin, and gums.
Approximately 60% to 70% of adults with AML can be expected to attain CR status following appropriate induction therapy. More than 25% of adults with AML (about 45% of those who attain CR) can be expected to survive 3 or more years and may be cured. Remission rates in adult AML are inversely related to age, with an expected remission rate of more than 65% for those younger than 60 years.
Patients with leukemias that express the progenitor cell antigen CD34 and/or the P-glycoprotein (MDR1 gene product) have an inferior outcome. AML associated with an internal tandem duplication of the FLT3 gene (FLT3/ITD mutation) has an inferior outcome that is attributed to a higher relapse rate.
FLT3 (Fms-Related Tyrosine Kinase; Stem Cell Tyrosine Kinase)
This gene encodes a class III receptor tyrosine kinase that regulates hematopoiesis. This receptor is activated by binding of the fms-related tyrosine kinase 3 ligand to the extracellular domain, which induces homodimer formation in the plasma membrane leading to autophosphorylation of the receptor. The activated receptor kinase subsequently phosphorylates and activates multiple cytoplasmic effector molecules in pathways involved in apoptosis, proliferation, and differentiation of hematopoietic cells in bone marrow. Mutations that result in the constitutive activation of this receptor result in acute myeloid leukemia and acute lymphoblastic leukemia.
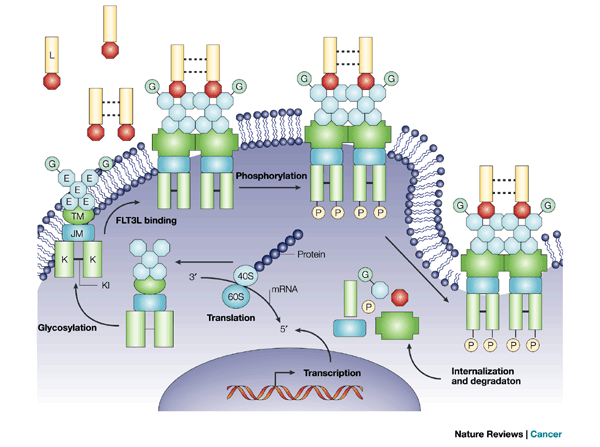
Transcription of the FMS-like tyrosine kinase 3 (FLT3) gene produces FLT3 mRNA, which is translated to FLT3 protein. FLT3 contains five extracellular immunoglobulin-like domains (E), a transmembrane domain (TM), a juxtamembrane domain (JM) and two tyrosine-kinase domains (K) that are linked through the tyrosine-kinase insert (KI). Cytoplasmic FLT3 undergoes glycosylation (G), which promotes localization of the receptor to the membrane. Wild-type FLT3 remains as a monomeric, inactivated protein on the cell surface until FLT3 ligand (L), probably in a dimeric form, binds the receptor and induces receptor dimerization. FLT3 dimerization promotes phosphorylation (P) of the tyrosine-kinase domains, thereby activating the receptor and downstream effectors. The dimerized receptors are quickly internalized and degraded. http://www.nature.com/nrc/journal/v3/n9/fig_tab/nrc1169_F2.html
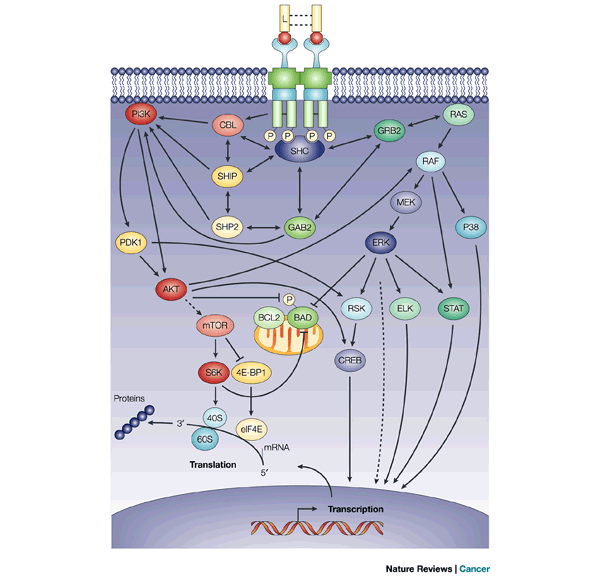
Although the FMS-like tyrosine kinase 3 (FLT3) signaling cascade has not been definitively characterized, this figure shows some of the complex associations and downstream effects that probably occur after activation of FLT3. Binding of FLT3 ligand (L) to FLT3 triggers the PI3K (phosphatidylinositol 3-kinase) and RAS pathways, leading to increased cell proliferation and the inhibition of apoptosis. PI3K activity is probably regulated through various interactions between FLT3, SH2-containing sequence proteins (SHCs) and one or more other proteins, such as SH2-domain-containing inositol phosphatase (SHIP), SH2-domain-containing protein tyrosine phosphatase 2 (SHP2), CBL (a proto-oncogene) and GRB2-binding protein (GAB2). Activated PI3K stimulates downstream proteins such as 3-phosphoinositide-dependent protein kinase 1 (PDK1), protein kinase B (PKB/AKT) and the mammalian target of rapamycin (mTOR), which initiate the transcription and translation of crucial regulatory genes through the activation of p70 S6 kinase (S6K) and the inhibition of eukaryotic initiation factor 4E-binding protein (4E-BP1). In addition, PI3K activation blocks apoptosis through phosphorylation of the pro-apoptotic BCL2-family protein BAD (BCL2 antagonist of cell death). Activated FLT3 also associates with GRB2 through SHC, so activating RAS. RAS activation stimulates downstream effectors such as RAF, MAPK/ERK kinases (MEKs), extracellular-signal-regulated kinase (ERK), and the 90-kDa ribosomal protein S6 kinase (RSK). These downstream effectors activate cyclic adenosine monophosphate-response element binding protein (CREB), ELK and signal transducer and activators of transcription (STATs), which lead to the transcription of genes involved in proliferation. Both pathways probably also interact with many other anti-apoptotic and cell-cycle proteins, such as WAF1, KIP1 and BRCA1. Solid arrows depict direct associations between proteins, whereas dashed arrows depict associations that have been indicated indirectly. http://www.nature.com/nrc/journal/v3/n9/fig_tab/nrc1169_F3.html
FLT3 mutations lead to constitutive activation
The FMS-like tyrosine kinase 3 (FLT3) is expressed on the cell surface of hematopoietic progenitors and its ligand can be found in either a soluble form or a membrane-bound form on bone marrow stromal cells. In 1996, it was reported that an internal tandem duplication (ITD) of base pairs within the juxtamembrane coding portion or point mutations in the second kinase domain (KDM) could result in constitutive activation of the gene in acute myeloid leukemia (AML) patients (see figure). These abnormalities are seen in up to 30% of patients with AML and especially in those with normal cytogenetics and with acute promyelocytic leukemia, making it one of the most frequently identified molecular abnormalities in AML.
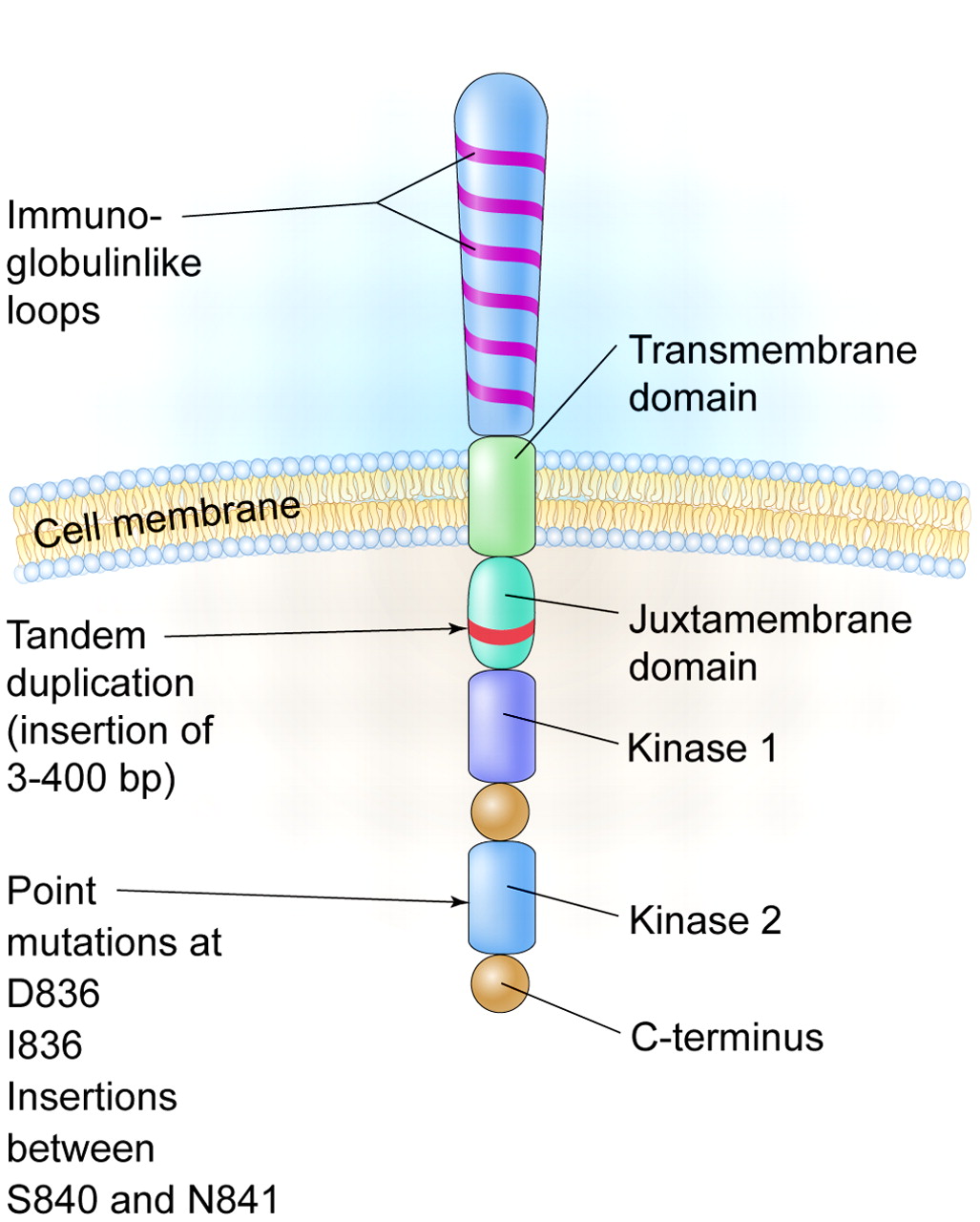
A schematic diagram of the FLT3 receptor tyrosine kinase showing the location of the internal tandem duplication of genes within the juxtamembrane domain and point mutations and gene insertions in the second kinase domain. http://www.bloodjournal.org/content/106/10/3331?sso-checked=true
Gilteritinib
Gilteritinib is a receptor tyrosine kinase inhibitor of FLT3 and AXL, which are involved in the growth of cancer cells. Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) as well as tyrosine kinase domain (TKD), two common types of FLT3 mutations that are seen in up to one third of patients with AML.
The gilteritinib Phase 3 trial follows a Phase 1/2 trial, which evaluated doses from 20 to 450 mg once daily. A parallel multi-dose expansion cohort was initiated based on the efficacy seen in the dose escalation phase. Preliminary data from the Phase 1/2 trial demonstrated a 57.5% overall response rate and a 47.2% composite Complete Response (CR) rate in 106 patients with FLT3 mutations who received 80 mg and higher doses. Median duration of response was 18 weeks across all doses and median OS was approximately 27 weeks at 80 mg and above in FLT3 mutation positive patients.
The new Phase 3 trial will enroll 369 patients with FLT3 activating mutation in bone marrow or whole blood AML who are refractory to or have relapsed after first-line AML therapy. Subjects will be randomised in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy consisting of LoDAC (low-dose cytarabine), azacitidine, MEC (mitoxantrone, etoposide, and intermediate-dose cytarabine), or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor with idarubicin).
What is AXL?
The protein encoded by this gene is a member of the Tyro3-Axl-Mer (TAM) receptor tyrosine kinase subfamily. The encoded protein possesses an extracellular domain which is composed of two immunoglobulin-like motifs at the N-terminal, followed by two fibronectin type-III motifs. It transduces signals from the extracellular matrix into the cytoplasm by binding to the vitamin K-dependent protein growth arrest-specific 6 (Gas6). This gene may be involved in several cellular functions including growth, migration, aggregation and anti-inflammation in multiple cell types. Alternative splicing results in multiple transcript variants of this gene.
AXL regulatess many physiological processes including cell survival, cell proliferation, migration and differentiation. Ligand binding at the cell surface induces dimerization and autophosphorylation of AXL. Following activation by ligand, ALX binds and induces tyrosine phosphorylation of PI3-kinase subunits PIK3R1, PIK3R2 and PIK3R3; but also GRB2, PLCG1, LCK and PTPN11. Other downstream substrate candidates for AXL are CBL, NCK2, SOCS1 and TNS2. Recruitment of GRB2 and phosphatidylinositol 3 kinase regulatory subunits by AXL leads to the downstream activation of the AKT kinase. GAS6/AXL signaling plays a role in various processes such as endothelial cell survival during acidification by preventing apoptosis, optimal cytokine signaling during human natural killer cell development, hepatic regeneration, gonadotropin-releasing hormone neuron survival and migration, platelet activation, or regulation of thrombotic responses. Plays also an important role in inhibition of Toll-like receptors (TLRs)-mediated innate immune response. In case of filovirus infection, seems to function as a cell entry factor.
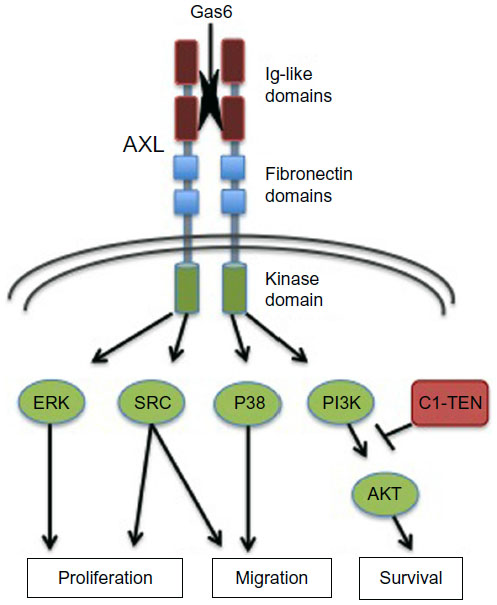
AXL structure and effector pathways. Shown are the primary structural determinants impacting AXL activation and signaling. The regions include the extracellular regions Ig-like and fibronectin domains important for ligand (Gas6) binding and receptor dimerization, as well as intracellular kinase and C-terminal docking regions critical for kinase activity and downstream signaling pathway engagement, as shown. AXL mediates a variety of cellular processes, as indicated. https://www.dovepress.com/axl-receptor-tyrosine-kinase-as-a-therapeutic-target-in-nsclc-peer-reviewed-fulltext-article-LCTT
AXL role in AML?
Shinh et al compared AXL expression in 58 patient-derived lung adenocarcinoma tissue specimens and identified ~48% (28/58) as positive and ~52% (30/58) with negative expression. AXL protein expression was retrospectively evaluated with clinicopathologic features and found to be significantly correlated with lymph node involvement (P<0.0001) and advanced clinical stage (P<0.0001). Further clinical outcome data in early-stage, resected lung adenocarcinomas demonstrated the prognostic impact of AXL expression on patient survival with a significant difference in 5-year overall survival rates comparing high and low AXL protein levels (38.6% and 77.5%, respectively; P<0.0001).
AXL represents an independent prognostic marker and therapeutic target in AML. AML cells induce expression and secretion of the AXL ligand growth arrest-specific gene 6 (Gas6) by bone marrow-derived stromal cells (BMDSCs). Gas6 in turn mediates proliferation, survival, and chemoresistance of AXL-expressing AML cells. This Gas6-Axl paracrine axis between AML cells and BMDSCs establishes a chemo-protective tumor cell niche that can be abrogated by AXL-targeting approaches. AXL inhibition is active in FLT3-mutated and FLT3 wild-type AML, improves clinically relevant end points, and its efficacy depends on presence of Gas6 and AXL. AXL inhibition alone or in combination with chemotherapy might represent a novel therapeutic avenue for AML.