

Question: How do you turn aggressive breast, lung, and bladder cancer cells back into benign cells?
Answer: microRNAi’s.
Researchers at the Mayo Clinic demonstrated that PLEKHA7 maintains the normal state of cells, via select miRNAs, by tethering the microprocessor involved in miRNA production to E-cadherin and p120, allowing the adhesion proteins to exert their tumour-suppressing effects. Conversely, “when this apical adhesion complex was disrupted after loss of PLEKHA7, this set of miRNAs was mis-regulated, and the E-cadherin and p120 switched sides to become oncogenic,” noted senior investigator Panos Anastasiadis.
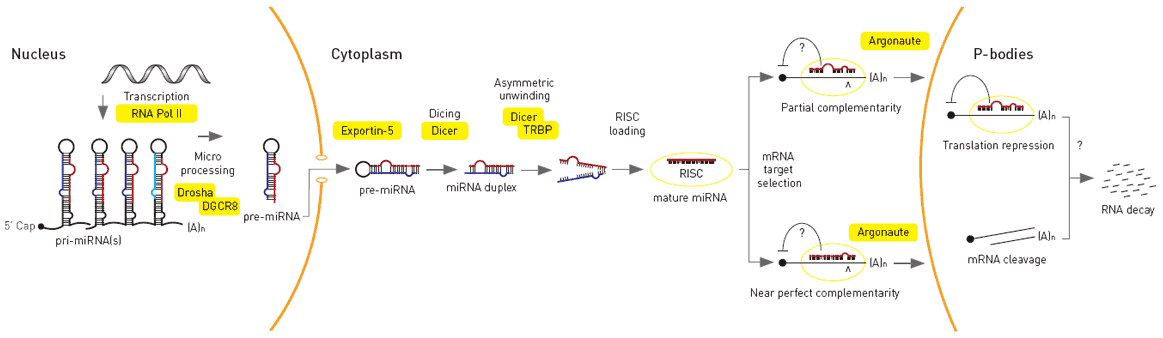
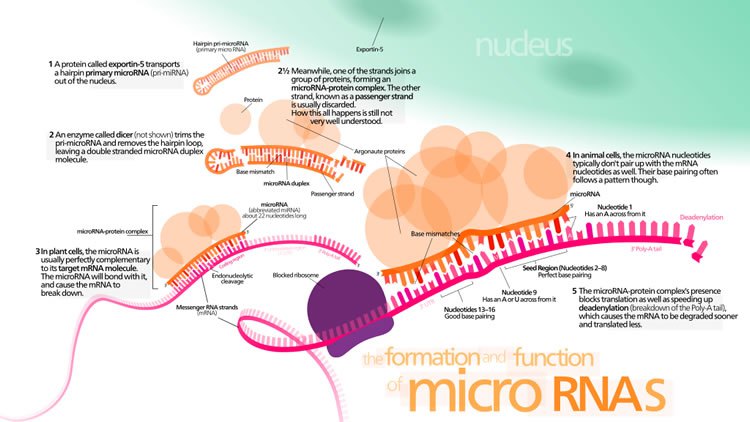
MicroRNA genes are transcribed by RNA polymerase II as large primary transcripts (pri-microRNA) that are processed by a protein complex containing the RNase III enzyme Drosha, to form an approximately 70 nucleotide precursor microRNA (pre-microRNA). This precursor is subsequently transported to the cytoplasm where it is processed by a second RNase III enzyme, DICER, to form a mature microRNA of approximately 22 nucleotides (Figure 1). The mature microRNA is then incorporated into a ribonuclear particle to form the RNA-induced silencing complex, RISC, which mediates gene silencing.
E-cadherin and p120 catenin (p120) are essential for epithelial homeostasis, but can also exert pro-tumorigenic activities. Here, we resolve this apparent paradox by identifying two spatially and functionally distinct junctional complexes in non-transformed polarized epithelial cells: one growth suppressing at the apical zonula adherens (ZA), defined by the p120 partner PLEKHA7 and a non-nuclear subset of the core microprocessor components DROSHA and DGCR8, and one growth promoting at basolateral areas of cell-cell contact containing tyrosine-phosphorylated p120 and active Src. Recruitment of DROSHA and DGCR8 to the ZA is PLEKHA7 dependent. The PLEKHA7-microprocessor complex co-precipitates with primary microRNAs (pri-miRNAs) and possesses pri-miRNA processing activity. PLEKHA7 regulates the levels of select miRNAs, in particular processing of miR-30b, to suppress expression of cell transforming markers promoted by the basolateral complex, including SNAI1, MYC and CCND1. Our work identifies a mechanism through which adhesion complexes regulate cellular behaviour and reveals their surprising association with the microprocessor.
SNAI1 is involved in the EMT (epithelial-mesenchymal switch). MYC is a transcription factor that drives loss of apoptosis, cell cycle progression, cellular transformation, and immortalization. CCND1 forms a complex with and functions as a regulatory subunit of CDK4 or CDK6, whose activity is required for cell cycle G1/S transition. By losing function of PLEKHA7 results in the loss of miRNA that block three critically important molecules that are responsible for the aggressive cancer phenotype. P120 regulates cell adhesion via its modulation of cadherins at the cell surface, and modulates the activities of RhoA, Rac, and Cdc42, suggesting that along with other Src substrates, p120 regulates actin dynamics. Without the PLEKHA7 microprocessor complex, p120 functions in a pro-oncogenic fashion that results in malignant transformation rather than a pro-homeostatic fashion.
“We believe that loss of the apical PLEKHA7-microprocessor complex is an early and somewhat universal event in cancer,” he adds. “In the vast majority of human tumor samples we examined, this apical structure is absent, although E-cadherin and p120 are still present. This produces the equivalent of a speeding car that has a lot of gas (the bad p120) and no brakes (the PLEKHA7-microprocessor complex).
Very interesting findings! Hopefully this can lead to more understanding of cancer and ways to “reprogram” cancerous cells.