Can-Fite BioPharma announced that it has dosed the first patient in a Phase II trial for the treatment of hepatocellular carcinoma (HCC), the most common form of liver cancer. The study will enroll 78 patients with refractory advanced hepatocellular carcinoma with Child-Pugh Class B cirrhosis. Patients will receive 25 mg of CF102 orally twice a day; the endpoint will be overall survival between those receiving CF102 versus placebo.
CF102 is a small molecule that binds to the Gi protein-associated A(3) adenosine receptor (A3AR), a member of the adenosine receptor family. Upon induction, the A3 adenosine receptor, A3AR, suppresses the formation of cAMP and its downstream effectors. Studies have indicated that activation of A3AR by its agonist, IB-MECA, results in growth inhibition of malignant cells. CF102 is an oral small molecule drug generically known as Cl-IB-MECA (2-chloro-N6-(3-iodobenzyl)-adenosine-5’- N-methyl-uronamide), a highly specific and selective agonist at the A3 adenosine receptor (A3AR)specific agonist of A3AR.
The Gi protein coupled A3 adenosine receptor (A3AR) is highly expressed in inflammatory and cancer cells whereas low expression is found in normal body cells. High A3AR expression levels are also found in peripheral blood mononuclear cells (PBMCs) of patients with inflammatory and cancer diseases, reflecting A3AR expression in the pathological remote sites.
Research further suggests that A3AR mediates a differential effect on pathological and normal cells. While specific A3AR agonists, such as CF101 and CF102, induce apoptosis of inflammatory and cancer cells, normal cells are refractory to the effects of the drug. This differential effect attributes to the safety profile of the A3AR agonists.
Mechanism of Action
Mechanistically, CF102 decreases the expression level of phosphorylated glycogen synthase kinase-3β, NF-κB, and TNF-α and prevented apoptosis in the liver. This was demonstrated by decreased expression levels of Fas receptor (FasR) and of the pro-apoptotic proteins Bax and Bad in liver tissues.
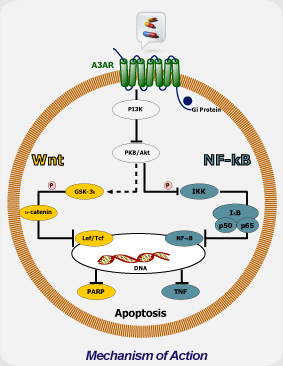
CF102 mechanism of action is mediated via de-regulation of the NF-κB and the Wnt signal transduction pathways, resulting in apoptosis of tumor cells. The protective effect of CF102 is mediated via down-regulation of the NF-κB signal transduction pathway and preventing apoptosis.
Apoptosis & NFkB
NFkB induces Inhibitors of apoptosis as well as Bcl-2 and Bcl-XL, which inhibit pro-apoptotic Bax and Bak proteins.
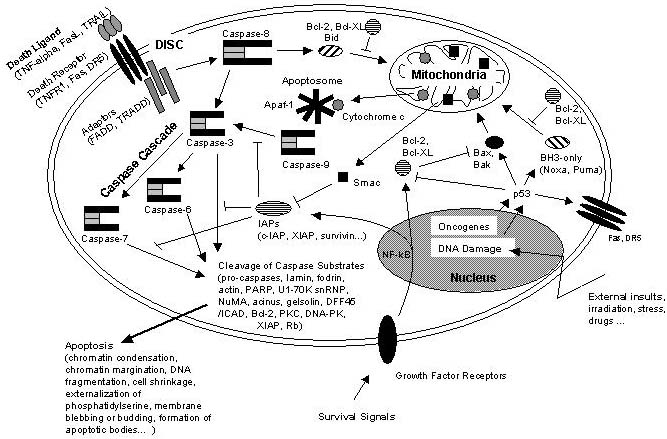
Schematic representation of some major apoptotic signalling pathways. Apoptosis can be induced in response to various signals from inside and outside the cell, e.g. by ligation of so called death receptors or by cellular stress triggered by oncogenes, irradiation or drugs. Signals emanating from death receptors initially activate the Death Inducing Signaling Complex (DISC) which mediates activation of the initiator caspase-8. Activated caspase-8 initiates a caspase cascade by processing the effector caspases-3, -6, and – 7 which in turn cleave a number of protein substrates. Cleavage of caspase substrates eventually leads to the characteristic morphological and biochemical features of apoptosis. In some cell systems, this direct caspase cascade is sufficient to elicit apoptosis on its own (type 1 signaling), whereas in other cases the signal coming from the DISC must be amplified by the proteolytic activation of the BH3-only protein Bid by caspase-8 with subsequent induction of apoptotic events at the mitochondria (type 2 signaling). Mitochondrial apoptotic signaling includes the release of cytochrome c from the mitochondrial intermembrane space to the cytosol where it contributes to the formation of the apoptosome which consists of cytochrome c, Apaf-1 and dATP. The apoptosome activates caspase-9 which is another initiator caspase and thus is able to mediate the caspase cascade by activating caspase-3. Another mitochondrial pro-apoptotic factor is Smac which acts by inhibiting the IAPs from blocking caspase activity. IAPs are a family of proteins with anti-apoptotic activity by directly inhibiting caspases. IAP expression can be upregulated in response to survival signals such as those coming from growth factor receptors, e.g. by activation of the transcription factor NF-kB, therefore providing a means to suppress apoptosis signaling. Of central importance are the anti-apoptotic Bcl-2 family members such as Bcl-2 and Bcl-XL which counteract the action of BH3-only proteins such as Bid but also of pro-apoptotic Bax and Bak and thus can inhibit mitochondrial pro-apoptotic events. Apoptotic signals coming from the inside of the cell frequently have their origin within the nucleus, being a consequence of DNA damage induced by irradiation, drugs or other sort of stress. DNA damage in most cases eventually results in the activation of the p53 transcription factor which promotes expression of pro-apoptotic Bcl-2 members and suppresses anti-apoptotic Bcl-2 and Bcl-XL. Other organelles besides mitochondria and the nucleus, such as the ER and lysosomes also have been implicated in apoptotic signaling pathways, and it should be kept in mind that presumably hundreds of proteins are part of an extremely fine-tuned regulatory network consisting of pro- and anti-apoptotic factors. http://www.celldeath.de/encyclo/aporev/revfigs/revfig_6.htm
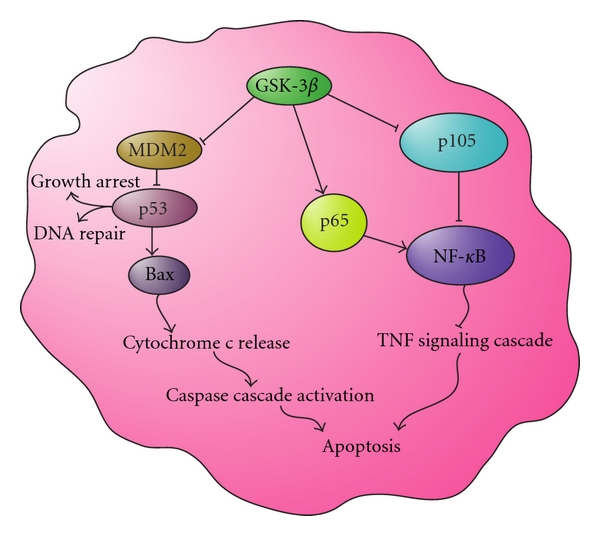
Apoptosis and GSK-3ß
Active GSK-3β inhibits MDM2 regulation of p53, leading to DNA repair and growth arrest, and in some cases the activation of the caspase cascade through Bax to promote apoptosis. Active GSK-3β also positively regulates NFκB by activating IKK, IκB, and p65, leading to the inhibition of TNF-mediated apoptosis. These actions inhibit the initiation of apoptosis through the TNF signaling cascade.