

The Ras onogene has been studied for 30 years, with over 40,000 scientific articles published on it. However, an inhibitor of the mutated form of Ras has not been able to be developed. So, researchers have developed downstream blockers of the Ras cascade – MEK inhibitors and Raf inhibitors, for example. But why can’t a molecule that blocks mutated Ras be identified?
The National Cancer Institute (NCI) last year announced a $10 million effort, dubbed the RAS Program, aimed at developing new ideas for targeting the protein. Frank McCormick, PhD, FRS, a leader in Ras research, recently stepped down as director of theUCSF Helen Diller Comprehensive Cancer Center to lead the new NCI project.
“A lot of people got into the drug discovery for Ras very early on and failed,” McCormick explained. “The clinical need [for a Ras drug] is the same or perhaps even better recognized than it was 30 years ago, but technology has advanced to the point where it’s worth having another shot at it.”
Recently, a small molecule that binds to a pocket in mutated Ras that accounts for 7% of lung cancer was identified. The molecule leaves un-mutated forms of Ras fully functional. Researchers at UCSF, led by Kevan Shokat, are optimizing the compound for development as a drug. But, this compound is for a particular mutation to K-ras; it does not inhibit other mutations to K-ras, or mutated N-ras or H-ras. (See – Targeting a Key Driver of Cancer.)
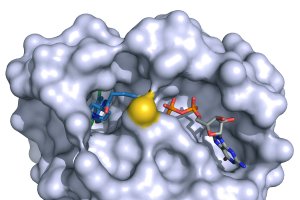
Molecule that binds to and mutated K-ras that occurs in 7% of lung cancers. It blocks the constitutive firing of this mutated K-ras.
The Ras pathway is the central signal transduction pathway in the cell, and mutations in this cascade (upstream or downstream) are responsible for driving many cancers. (See – Ras signaling pathway in GeneCopoeia.)
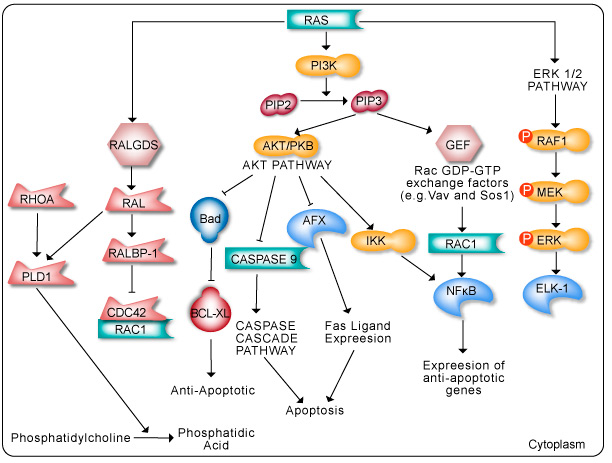
The RAS family of GTPases (HRAS, NRAS, and KRAS) comprises proteins that are highly conserved across species and has key roles in numerous basic cellular functions, including control of proliferation, differentiation, and apoptosis. Many of these Ras mediated signals are interpreted differently depending on the cell type or microenvironment receiving the stimulus. Not all of these effectors are activated in any given cell type. The first RAS effector pathway identified was the RAF-MEK-ERK pathway. This pathway is an essential, shared element of mitogenic signaling involving tyrosine kinase receptors, leading to a wide range of cellular responses, including growth, differentiation, inflammation, and apoptosis. The RAF family of proteins (Raf-1, A-Raf, and B-Raf) is serine/threonine kinases that bind to the effector region of RAS-GTP, thus inducing translocation of the protein to the plasma membrane. The second best-characterized RAS effector family is phosphoinositide 3-kinases (PI3Ks). PIP3 stimulates the AKT/PKB kinase and several of the Rac-GEF’s such as Sos1 and Vav. AKT activation inhibits apoptosis by inhibiting the actions of Bad, Caspase 9 and AFX. AKT further hinders apoptosis by phosphorylating the IkB repressor of NFkB. This pathway plays important roles as mediators of RAS-mediated cell survival and proliferation.
Mutations in all isoforms or Ras result in Ras-GTP unable to be hydrolyzed to Ras-GDP, thereby rendering Ras constitutively active. (See – A Comprehensive Survey of Ras Mutations in Cancer.)
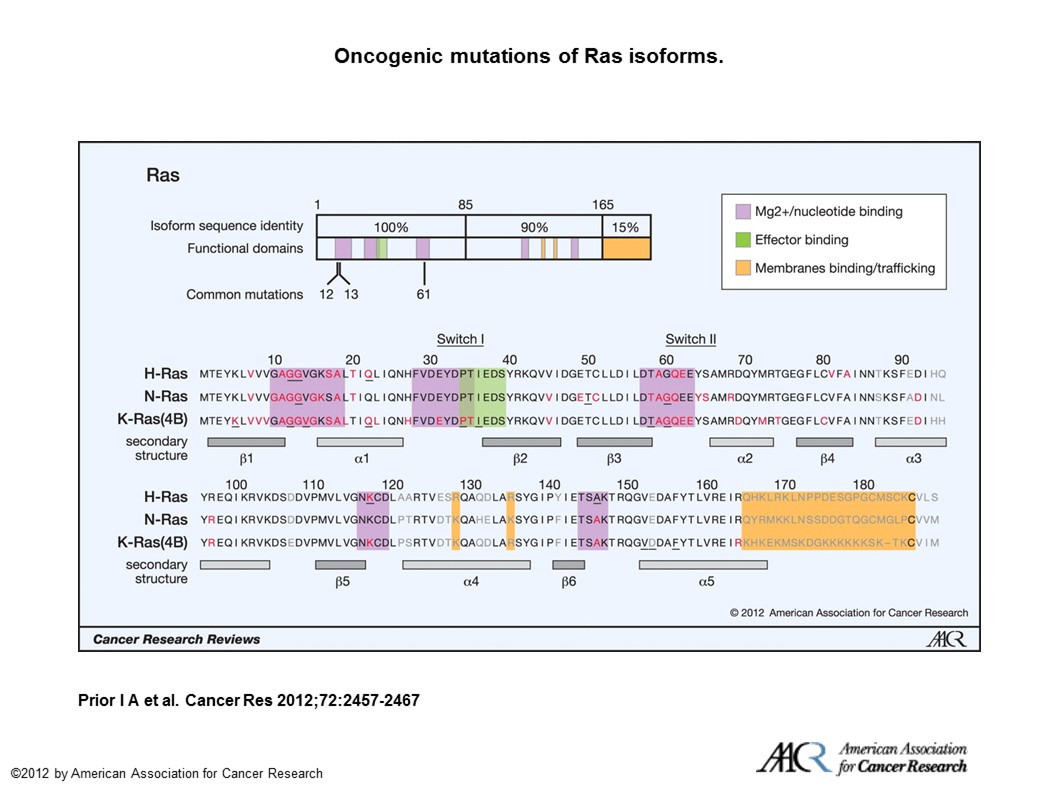
Oncogenic mutations of Ras isoforms. The key oncogenic mutations are in the region that is identical among the 3 isoforms. Forty-four separate point mutations have been characterized in Ras isoforms, with 99.2% of all mutations occurring at codons 12, 13, and 61. Mutations that cluster in and around loops 1, 2, and 4 are responsible for nucleotide binding and result in enhanced GTP binding. Residues that are mutated in cancer are highlighted in red, those that are mutated in developmental disorders are underlined, and those that are variable among isoforms are in gray (26, 65, 66).