
NQO1 [NAD(P)H:quinone oxidoreductase 1 over-expression has been shown to confer a poor prognosis for patients with cancer of the breast, colon, cervix, lung and pancreas. Recent research indicates that the mechanism involves stabilization HIF-1α (hypoxia inducible factor-1) such that it is not degraded.
What is HIF-1α?
HIF-1α is a transcription factor whose activity is regulated by intracellular oxygen levels. Under normal conditions (normoxia), hydroxylation of proline residues leads to binding with VHL (von Hipel-Landau) complex and proteasomal degradation. Thus, under normal conditions, it is produced to be degraded, but immediately available should the cell come under hypoxic stress. Under hypoxic conditions, proline residues are not hydroxylated and HIF-1α translocates to the nucleus activating many genes, including the expression of VEGF (vascular endothelial factor) to promote angiogenesis to increase oxygen supply to the cell.
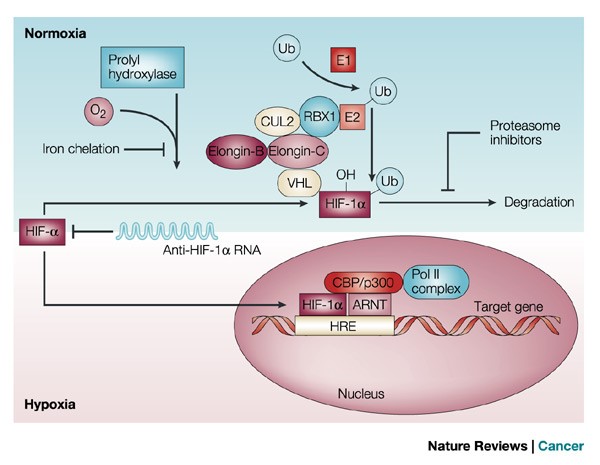
Figure 1. In the presence of oxygen (O2), prolyl hydroxylase post-translationally modifies hypoxia-inducible transcription factor (HIF)-1α, allowing it to interact with the von Hippel–Lindau (VHL) complex. Prolyl hydroxylase contains an iron moiety, so iron chelation inhibits this activity. VHL is part of a larger complex that includes elongin-B, elongin-C, CUL2, RBX1 and a ubiquitin-conjugating enzyme (E2). This complex, together with a ubiquitin-activating enzyme (E1), mediates the ubiquitylation (Ub) of HIF-1α. The Ub modification targets HIF-1α for degradation, which can be blocked by proteasome inhibitors. In the absence of oxygen, prolyl hydroxylase cannot modify HIF-1α, and the protein remains stable. Stabilized HIF-1α is translocated to the nucleus, where it interacts with cofactors such as aryl hydrocarbon receptor nuclear translocator (ARNT), CBP/p300 and the DNA polymerase II (Pol II) complex to bind to hypoxia-responsive element (HREs) and activate transcription of target genes97. ARNT2 and MOP3 are other proteins that have been shown to heterodimerize with HIF-1α (not shown). A natural HIF-1α antisense mRNA has been found in renal cancer. http://www.nature.com/nrc/journal/v2/n1/fig_tab/nrc704_F1.html
Why is HIF-1α important in cancer pathogenesis?
In heart disease and COPD, HIF-1α mediated changes are beneficial to us. Not so in cancer. As cancer cells divide, the distance from blood vessels increases causing hypoxic conditions in the tumor microenvironment. Moreover, as the cancer cell increases in mass, it compresses the blood vessels that are in its vicinity, thereby worsening the hypoxic conditions.
Tumor cells in this hypoxic region begin to adapt these low oxygen tension conditions by activating several survival pathways. Activation of HIF-1α transcription factor is the most recognized pathway adopted by hypoxic cells in this harsh microenvironment.
Many recent studies have provided convincing evidences of strong correlation between elevated levels of HIF-1α and tumor metastasis, angiogenesis, poor patient prognosis as well as tumor resistance therapy. It was found that hypoxia (low O2 levels) is a common character in many types of solid tumors. As an adaptive response to hypoxic stress, hypoxic tumor cells activate several survival pathways to carry out their essential biological processes in different ways compared with normal cells. Recent advances in cancer biology at the cellular and molecular levels highlighted the HIF-1α pathway as a crucial survival pathway for which novel strategies of cancer therapy could be developed. However, targeting the HIF-1α pathway has been a challenging but promising progresses have been made in the past twenty years. This review summarizes the role and regulation of the HIF-1α in cancer, and recent therapeutic approaches targeting this important pathway.
Activated HIF-1α plays a crucial role in adaptive responses of the tumor cells to changes in oxygen through transcriptional activation of over 100 downstream genes which regulate vital biological processes required for tumor survival and progression. Examples include genes involved in glucose metabolism, cell proliferation, migration and angiogenesis (Fig. 1). For example, in a rapidly growing tumor tissue, HIF-1α helps hypoxic tumor cells to shift glucose metabolism from the more efficient oxidative phosphorylation to the less efficient glycolytic pathway in order to maintain their energy production (the Warburg effect). For this reason, hypoxic cells tend to consume more glucose in order to meet their energy needs. HIF-1α mediates this metabolic conversion through the induction of enzymes involved in the glycolysis pathway and overexpression of glucose transporters (GLUTs) which increase glucose import into tumor cells . Another example involves the HIF-1α transcriptional induction of several pro-angiogenic factors such as the vascular endothelial growth factor (VEGF), which in turn stimulates the development of new blood vessels to enrich tumor cells with oxygen for their growth. In addition, HIF-1α promotes tumor metastasis into distant and more oxygenated tissues through the transcriptional activation of oncogenic growth factors such as transforming growth factor beta3 (TGF-β3), epidermal growth factor (EGF) and others. Altogether, HIF-1α activation in tumor cells is one of the key masters orchestrating their adaptation mechanisms to the hypoxia environment.
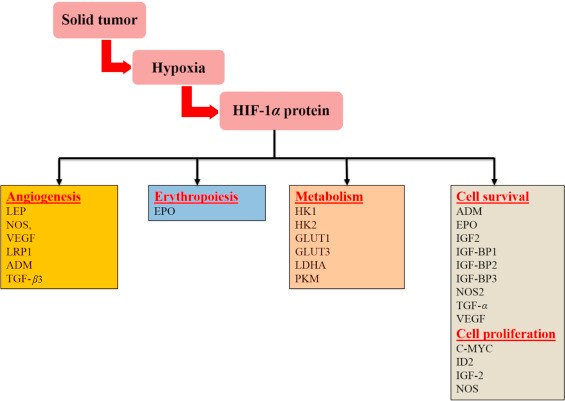
Figure 2. Representative HIF-1α regulatory genes and their effects on cancer progression. LEP, leptin; NOS, nitric oxide synthase; VEGF, vascular endothelial growth factor; LRP1, LDL-receptor-related protein 1; ADM, adrenomedullin; TGF-β3, transforming growth factor-β3; EPO, erythropoietin; HK1, hexokinase 1; HK2, hexokinase 2; GLUT1, glucose transporter 1; GLUT3, glucose transporter 3; LDHA, lactate dehydrogenase; PKM, pyruvate kinase M; IGF2, insulin-like growth factor 2; IGF-BP2, IGF-factor-binding protein 2; IGF-BP3, IGF-factor-binding protein 3; TGF-α, transforming growth factor α; C-MYC, myelocytomatosis virus oncogene cellular homolog; ID2, DNA-binding protein inhibitor. http://www.sciencedirect.com/science/article/pii/S2211383515000817
How is HIF-1α been targeted in cancer drug development?
Given the critical importance of HIF-1α to the survival of cancer cells, much time and effort has been expended in targeting this pathway in the development of cancer therapies.
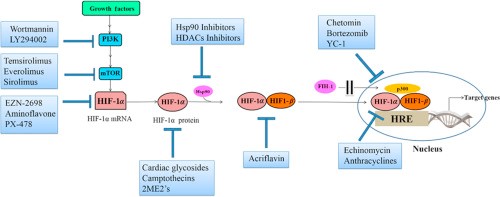
Back to NQO1
NQO1 has recently been shown to protect HIF-1α from proteasomal degradation. Thus, it permits HIF-1α translocation to the nucleus in transduced cells and activation of many pathways that lead to cancer progression.
NQO1 is a cytosolic reductase which is upregulated in many human cancers, including colorectal cancer, lung cancer, gastric cardiac carcinoma, melanoma, cholangiocarcinoma, pancreatic cancer, uterine cervical cancer and breast cancer. In breast, colorectal and cervical cancers, the high-level expression of NQO1 is closely associated with the late clinical stage, poor differentiation and lymph node metastasis. Consistently, breast and cervical cancer patients with high-level NQO1 expression have shown lower disease-free survival and 5-year overall survival rates compared with those with low-level NQO1 expression. Most canonical HIF-1α target genes (CA9, LDHA, GLUT1, PDK1, PGK1 and vascular endothelial growth factor (VEGF)) were significantly elevated in high NQO1-expressing tumours as compared with those with absent or low NQO1 expression, indicating that HIF-1α transcriptional signaling is accordingly activated in NQO1-expressing colorectal cancers. These results suggest that aberrantly elevated NQO1 expression correlates with the expression of HIF-1α and poor prognosis in colorectal cancer patients.
To investigate the NQO1-mediated activation of HIF-1α signalling at the cellular level, we further assessed HIF-1α expression in different cell types with NQO1 gain-of as well as loss-of-function. First, we generated RKO human colorectal cancer cells that express short hairpin RNA (shRNA) targeting NQO1 (RKO/pshNQO1) and the control scramble shRNA (RKO/pshCont). When the RKO/pshNQO1 cells were exposed to hypoxia (0.5% O2), the expression of HIF-1α was markedly decreased as compared with control RKO/pshCont cells. Immunocytochemical staining consistently showed that NQO1 enhances HIF-1α expression under hypoxia. We also exposed these cells to various oxygen tensions; 0.5, 1, 2, 5 or 20% O2, and we found that the knockdown of NQO1 significantly attenuates HIF-1α expression under mild hypoxia. We next investigated the NQO1 gain-of-function in MDA-MB-231 human breast cancer cells that are deficient in NQO1 expression. Overexpression of NQO1-myc-His6 (MDA-MB-231/pNQO1) dramatically increased the HIF-1α expression in both 20 and 0.5% O2. To further confirm that HIF-1α expression is regulated by NQO1, we transiently expressed various amount of pshNQO1 and pNQO1-myc-His6 in RKO and MDA-MB-231, respectively. Consistently, HIF-1α expression correlates with the levels of NQO1 expression. Ten human colorectal cancer cell lines and other types of cancer cell lines including HeLa (cervix), A549 (lung), PC3 (prostate) and U87-MG (brain) exhibited similar NQO1-mediated HIF-1α induction in both normoxia and hypoxia. To exclude the possibility of off-site effects of NQO1 shRNA, NQO1 expression was restored by overexpressing shRNA-resistant NQO1 in RKO/pshNQO1 cells. Reintroduction of NQO1 expression fully rescued the HIF-1α expression. In addition, immortalized mouse embryonic fibroblasts null for NQO1 (NQO1−/−) displayed markedly reduced levels of HIF-1α expression. We next sought to determine if NQO1 oxidoreductase enzymatic activity is necessary for HIF-1α induction. Introduction of point mutation at the known catalytic active site (C609T) that inactivates reductase activity had no effect on HIF-1α expression as compared with wild-type NQO1 indicating that NQO1 reductase enzymatic activity is not required for its HIF-1α inducing function. Taken together, these results clearly suggest that NQO1 has a crucial role in HIF-1α expression independent of its reductase function.
How does NOQ1 protect HIF-1α and what is the in vivo evidence?
NOQ1 binds to HIF-1α in the cytosol at amino acid residues 331 to 575, which contain the oxygen-dependent degradation (ODD) domain of HIF-1α. This prevents proteasomal degradation of HIF-1α.
Previous studies have shown that NQO1 stabilizes numerous proteins through inhibiting ubiquitination and the 26S proteasomal degradation. To assess if NQO1 binding to the ODD domain of HIF-1α accounts for the stabilization of HIF-1α via inhibiting ubiquitination and 26S proteasomal degradation, we measured the levels of ubiquitinated HIF-1α in cells with or without NQO1 expression. NQO1 knockdown (RKO/pshNQO1) and NQO1-deficient (MDA-MB-231) cells exhibited higher levels of ubiquitination in HIF-1α, whereas cells expressing high levels of NQO1 (RKO/pshCont and MDA-MB-231/pNQO1) suppressed ubiquitination in both hypoxia and normoxia. To further investigate whether NQO1 inhibits HIF-1α degradation via the ubiquitin-mediated proteasomal pathway under hypoxia, we measured HIF-1α protein levels after treating cells with cycloheximide and/or MG132 (a peptide aldehyde that blocks the proteolytic activity of proteasome 26s). MG132 treatment significantly inhibited the degradation of HIF-1α in NQO1-deficient cells. Collectively, these results indicate that NQO1 prevents HIF-1α from the ubiquitin-mediated proteasomal degradation.
To determine the functional contribution of NQO1 to HIF-1α expression and tumor growth in vivo, we evaluated the growth rate of NQO1-knockdown or overexpressing RKO xenografts in female BALB/c nude mice. NQO1 knockdown tumours displayed dramatically reduced growth rate, whereas tumours overexpressing NQO1 grew much faster as compared with control tumours. We assessed the levels of NQO1 and HIF-1α in these tumours and found that NQO1 knockdown tumours showed significantly reduced HIF-1α expression, which was associated with decreased proliferation (Ki67) and elevated apoptosis (cleaved caspase 3, CC3). In sharp contrast, NQO1 overexpressing tumours showed accelerated growth rate with higher cellular proliferation. We further assessed HIF-1α expression in relation to hypoxic areas by co-staining with hypoxia marker, pimonidazole, and HIF-1α. Control RKO tumours exhibited clear correlative HIF-1α expression along hypoxic areas. However, HIF-1α-positive areas were substantially expanded over the pimonidazole-positive hypoxic areas in NQO1-overexpressing tumours (RKO/pNQO1). Conversely, NQO1-knockdown tumours (RKO/pshNQO1) showed significantly less HIF-1α expression even in the pimonidazole-positive hypoxic areas. To confirm that this reduction in tumor growth is specifically due to a lack of HIF-1α stabilization by NQO1, rather than through other NQO1 mediated effects, we studied the growth rate of control (RKO/pshCont1/pshCont2), NQO1 overexpressing (RKO/pNQO1/pshCont2), and NQO1 overexpressing but HIF-1α knockdown (RKO/pNQO1/pshHIF-1α) xenograft tumours. Consistently, HIF-1α-knockdown almost completely inhibited tumor growth, even in the presence of overexpressed NQO1. This suggests that tumor growth is inhibited specifically through reduced NQO1-dependent HIF-1α expression. We further validated these results by quantifying Ki67 and CC3 expression and found that in accordance with our previous results, decreased HIF-1α expression in RKO tumours was associated with decreased proliferation (Ki67) and elevated apoptosis (CC3). Intriguingly, we found that NQO1 overexpression enhanced microvessel density while there was no difference in vascular density between control and HIF-1α knockdown tumours, measured using CD34 staining. Collectively, these observations strongly argue for the functional contribution of NQO1 in stabilizing HIF-1α which, in turn, enhances tumor growth in vivo.