
Adoptive immunotherapy with engineered T-cells, for example, CAR T-cells (Chimeric Antigen Receptor) is associated with significant toxicities including cytokine release syndrome, neurologic toxicity, “on target/off tumor” recognition, and anaphylaxis. CAR T-cells lack the ability to respond to signals that maintain immune homeostasis. For this reason, they are effective relentless killers of cells that express the target to which they have been programmed, however, at a safety cost.
A first-generation CAR typically comprises an antigen-binding ectodomain of a monoclonal antibody (mAb)-derived single chain variable fragment (scFv) and a signaling endodomain from the CD3ζ molecule, which contains 3 tyrosine kinase motifs that transmit activation signals. Following expression, the CAR re-directs the specificity of the T cell toward the cognate antigen. Unlike TCR, CAR are independent of MHC-restricted antigen presentation, which is a feature of immune evasion by many different cancers (Seliger et al., 1996; Seliger, 2008), and instead can bind directly to TAA (tumor associated antigen), albeit those located at the cell surface. They can also include multiple intracellular signaling domains from costimulatory molecules. For example, second-generation CAR may contain CD3ζ and CD28 signaling domains, while third-generation CAR may contain CD3ζ, CD28 and either OX40 (CD134) or 4-1BB (CD137) – see Figure 1. T cells expressing these later generation CAR demonstrate enhanced activation and effector function (Pule et al., 2005).
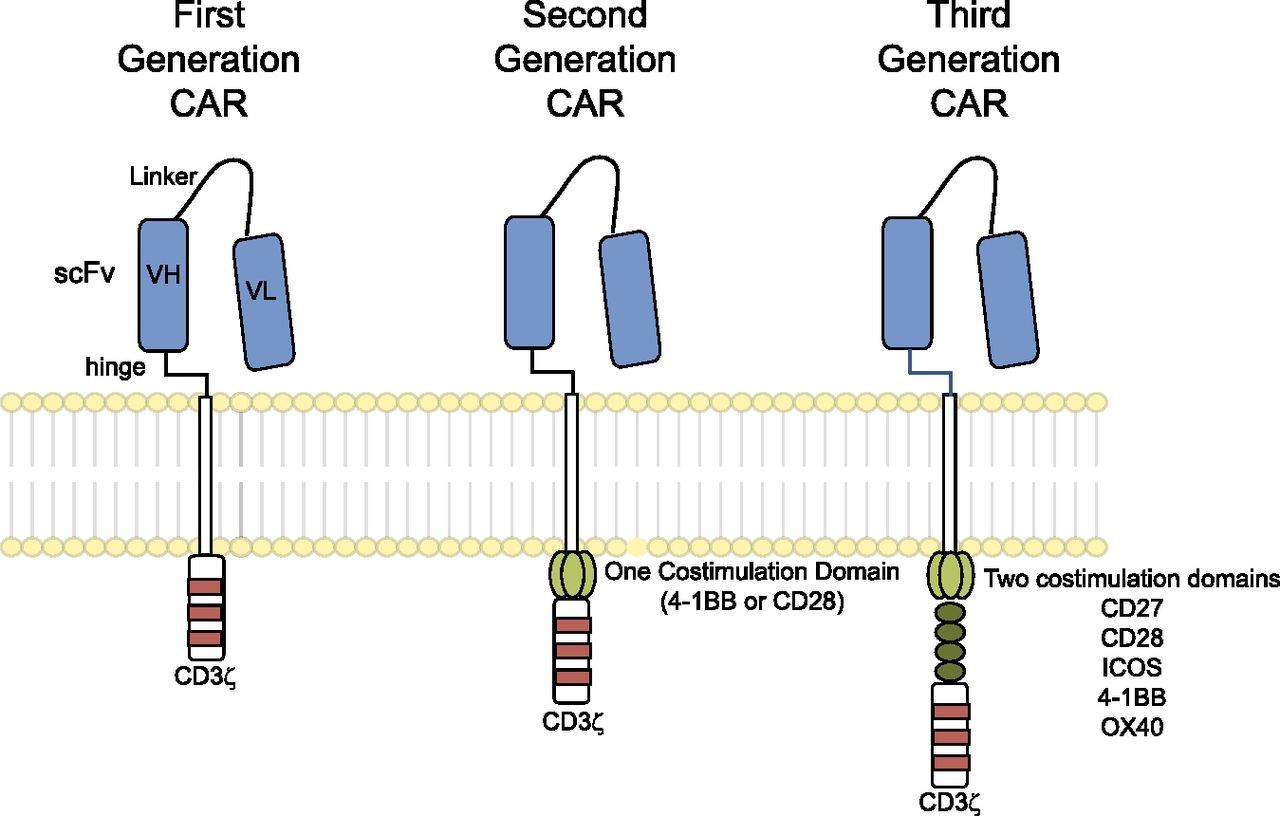
Figure 1. Chimeric antigen receptors. CARs target surface antigens in an MHC-independent fashion and consist of an ectodomain, hinge domain, transmembrane domain, and endodomain. The initial trials tested first-generation CARs that have a single cytoplasmic domain. Current trials are testing second- and third-generation CARs that have combinations of signaling domains. http://www.bloodjournal.org/content/123/17/2625?sso-checked=true
Autologous CAR T cells have shown promising objective responses in leukemia and neuroblastoma patients, including some complete and partial regressions (Pule et al., 2008; Porter et al., 2011; Grupp et al., 2013). However, reports of serious adverse events (AEs) in some of these early trials have raised safety concerns about the technology (Table 1).

Table 1. Reports of life-threatening and fatal adverse events in CAR T-cell clinical trials. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4211380/
Controlling CAR T-cells after they have achieved their desired mission, or when they go rogue and attack normal tissue, is the main focus of next generation CAR T-cell development. Three approaches are in development (Figure 2).
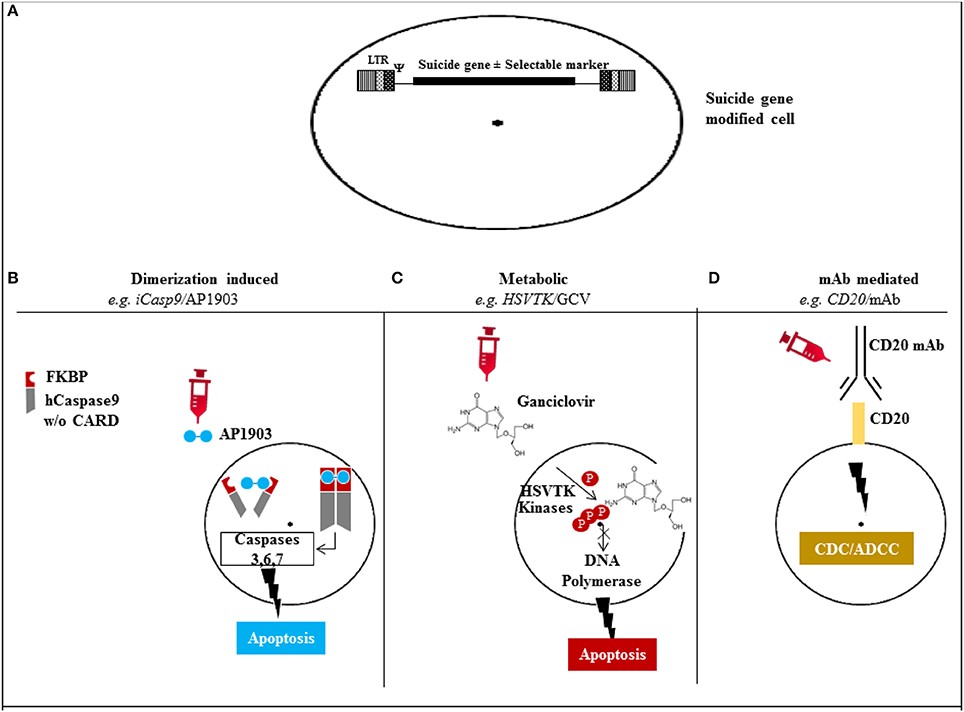
Figure 2. Mechanism of action of the different suicide gene technologies. (A) Suicide gene modification of cells of interest to allow conditional elimination in case of serious adverse events. Surface marker suicide genes, e.g., CD20, can also function as a selectable marker. (B) Dimerization induced e.g., iCasp9 protein with FKBP12-F36V binding domain joined to human caspase-9. Administration of AP1903 leads to dimerization of iCasp9 activating the intrinsic mitochondrial apoptotic pathway. (C) Metabolic, e.g., HSV/TK leads to phosphorylation of ganciclovir, and its triphosphate form (phosphorylated also through cellular kinases) incorporates into DNA with chain termination. (D) Monoclonal antibody (mAb) mediated, e.g., CD20 overexpression allows elimination after exposure to CD20 mAb through complement/antibody dependent cellular cytotoxicity (CDC/ADCC). LTR: long terminal repeat, psi: retroviral packaging element, iCasp9: inducible Caspase9, CARD: Caspase recruitment domain, HSVTK: herpes simplex virus thymidine kinase, GCV: ganciclovir, mAb: monoclonal antibody. http://journal.frontiersin.org/article/10.3389/fphar.2014.00254/full
Herpes Simplex Virus Thymidine Kinase (HSV-TK) Suicide Gene
Ganciclovir (GCV) is an acyclic nucleoside analogue of 2′-deoxyguanosine that inhibits replication of herpes viruses. The herpes simplex virus thymidine kinase (HSV-TK) phosphorylates ganciclovir to GCV-triphosphate, which is then incorporated into DNA, inhibiting DNA synthesis and resulting in cell death. Ganciclovir lacks deoxyribose, so, once it is incorporated into a growing DNA chain, elongation stops because nucleoside triphosphates cannot be added (Figure 3).
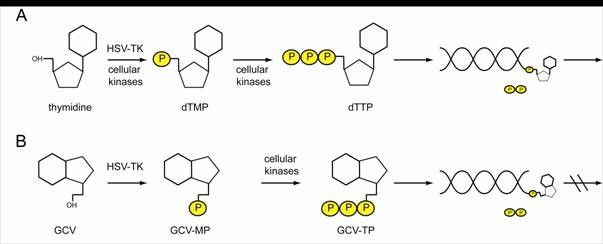
Figure 3. HSV-TK can phosphorylate thymidine and nucleotide analogs. (A) Thymidine is phosphorylated by cellular kinases or HSV-TK to thymidine-mono-phosphate (dTMP) and further to thymidine-tri-phosphate (dTTP) which can be incorporated into a replicating DNA strand. To the free OH- group, further nucleotides can be attached. (B) HSV-TK also phosphorylates nucleotide analogs like GCV, which in their tri-phosphorylated form are integrated in the newly synthesized DNA. As no further nucleotides can be attached to GCV, the replication process is discontinued. http://edoc.hu-berlin.de/dissertationen/kieback-elisa-2008-10-20/HTML/chapter1.html
Transfecting the gene for the thymidine kinase of herpes simplex virus into CAR T-cells provides a kill switch that can be activated later by the administration of ganciclovir, which is an approved drug for the treatment of HSV infection.
iCasp9 Suicide Gene
iCasp9 offers an alternative to HSV-TK; it consists of an inducible caspase 9 (iCasp9) gene together with the small-molecule, bio-inert, chemical induction of dimerization (CID) drug, AP1903. The iCasp9 gene contains the intracellular portion of the human caspase 9 protein, a pro-apoptotic molecule, fused to a drug-binding domain derived from human FK506-binding protein. Intravenous administration of AP1903 produces cross-linking of the drug-binding domains of this chimeric protein, which in turn dimerizes caspase9 and activates the downstream executioner caspase 3 molecule, resulting in cellular apoptosis (see Figures 4 and 5).
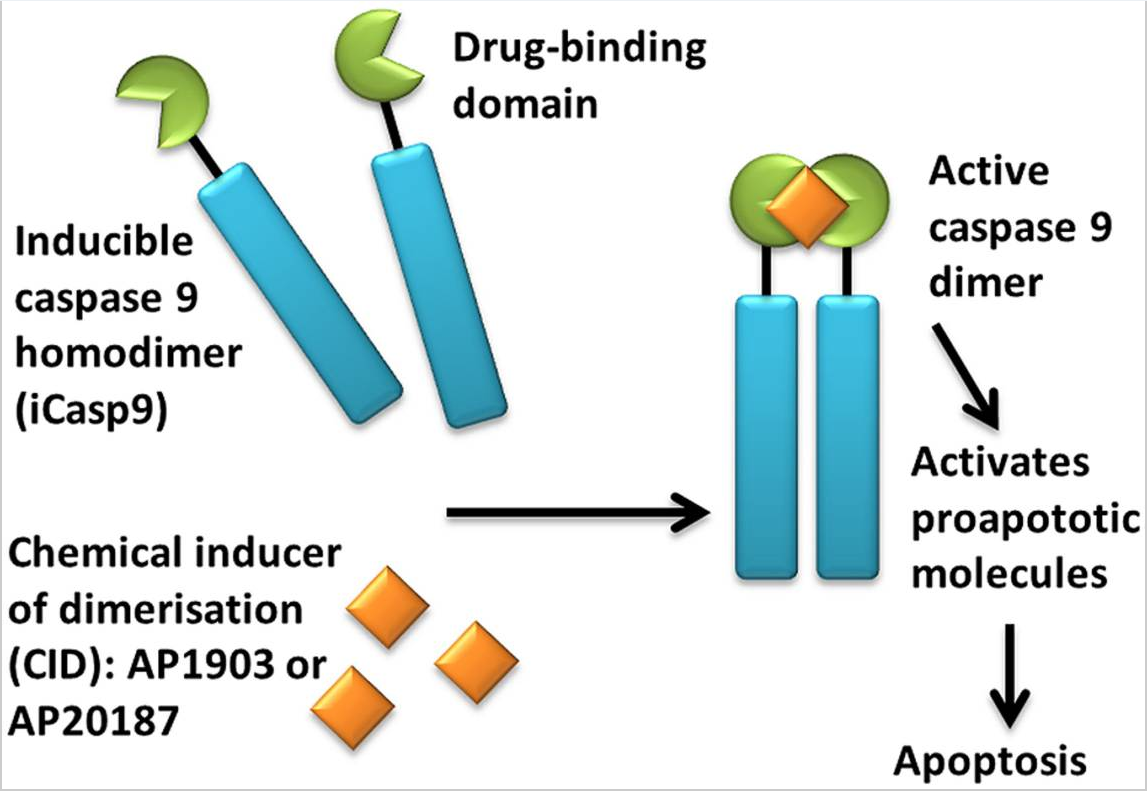
Figure 4. Activation of iCasp9 results in the death of transduced cells. Inducible caspase 9 (iCasp9) is produced in the transduced cell as a homodimer with a drug-binding domain. Administration of CID (AP1903 or AP20187) results in dimerization of caspase 9 leading to an activated form of the molecule, which then initiates a signaling cascade leading to apoptosis of the transduced cell. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4211380/
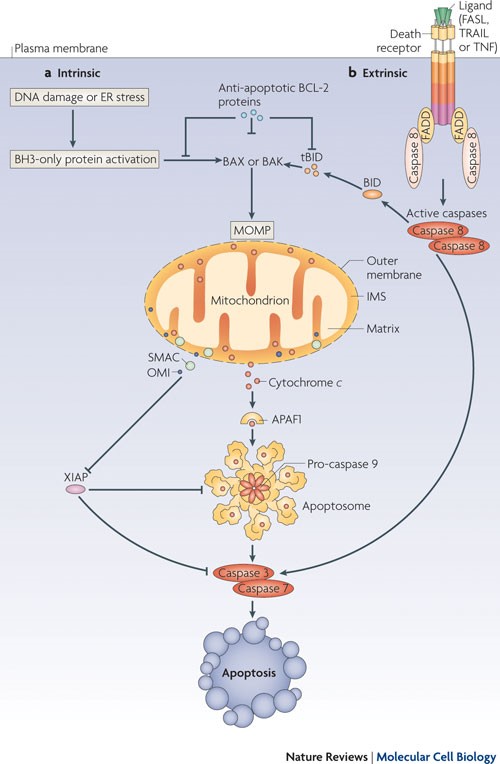
Figure 5. Caspase 9 cleaves and activates executioner caspases, caspase 3 and caspase 7, leading to apoptosis. http://www.nature.com/nrm/journal/v11/n9/fig_tab/nrm2952_F1.html
Extracellular Membrane Targets for Monoclonal Antibodies
Modification of CAR T-cells with cell surface antigens against which monoclonal antibodies are available, for example CD20 (a B-cell antigen) or EGFR (epidermal growth factor receptor). The administration of antibodies that bind to these extracellular membrane targets induces CAR T-cell death via opsonization followed by ADCC (antibody-dependent cell-mediated cytotoxicity) and CDC (complement-dependent cytotoxicity) – see Figures 6 and 7.
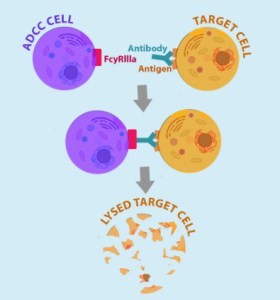
Figure 6. ADCC cell (macrophages and NK – natural killer cells) binds to Fc receptor portion of antibodies bound to the target cells (engineered CAR T-cells expressing cell surface antigen to which Fab portion of administered antibodies bind). http://being-bioreactive.com/2016/10/14/how-to-measure-antibody-dependent-cell-mediated-cytotoxicity-adcc/
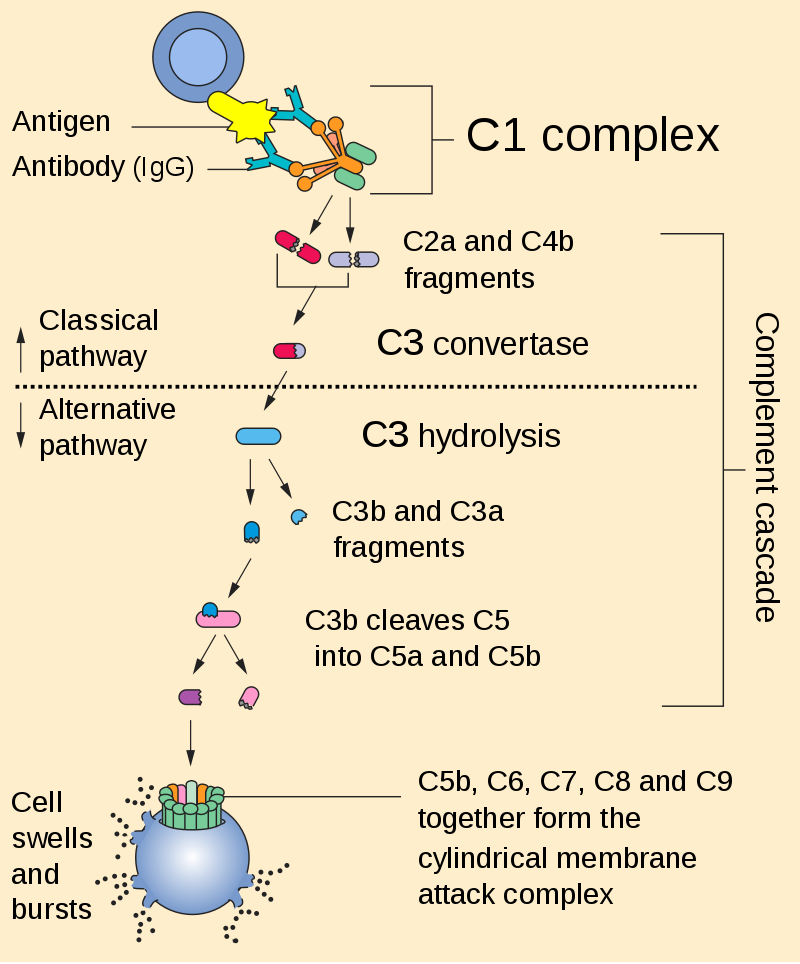
Figure 7. Complement cascade is initiated by the binding of the C1 complex to the Fc portion of the antibody that binds to the antigen expressed on the engineered CAR T-cell. https://en.wikipedia.org/wiki/Complement_system#/media/File:Complement_pathway.svg