
BRCA1/2 are DNA repair enzymes that are the essential components of homologous repair (HR), which is invoked when DNA doubles strand breaks are encountered. HR works via consultation of sister chromatids during late S to early G2 of the cell cycle – without repair of double strand breaks, mitotic catastrophe ensues:
Failure to arrest the cell cycle before or at mitosis triggers an attempt of aberrant chromosome segregation, which culminates in the activation of the apoptotic default pathway and cellular demise. Cell death occurring during the metaphase/anaphase transition is characterized by the activation of caspase-2 (which can be activated in response to DNA damage) and/or mitochondrial membrane permeabilization with the release of cell death effectors such as apoptosis-inducing factor and the caspase-9 and-3 activator cytochrome c.
How do PARP inhibitors work in the context of BRCA1/2 mutations?
PARP inhibitors are very effective in the treatment of ovarian and breast cancers with BRCA1/2 mutations; they work by blocking base excision repair (BER) and microhomologous mediated end-joining (MMEJ), which leads to stalled replication forms and doubles strand breaks. In patients without functioning BRCA1/2, PARP inhibitors overwhelm the cell with double strand breaks leading to mitotic catastrophe and cell death.
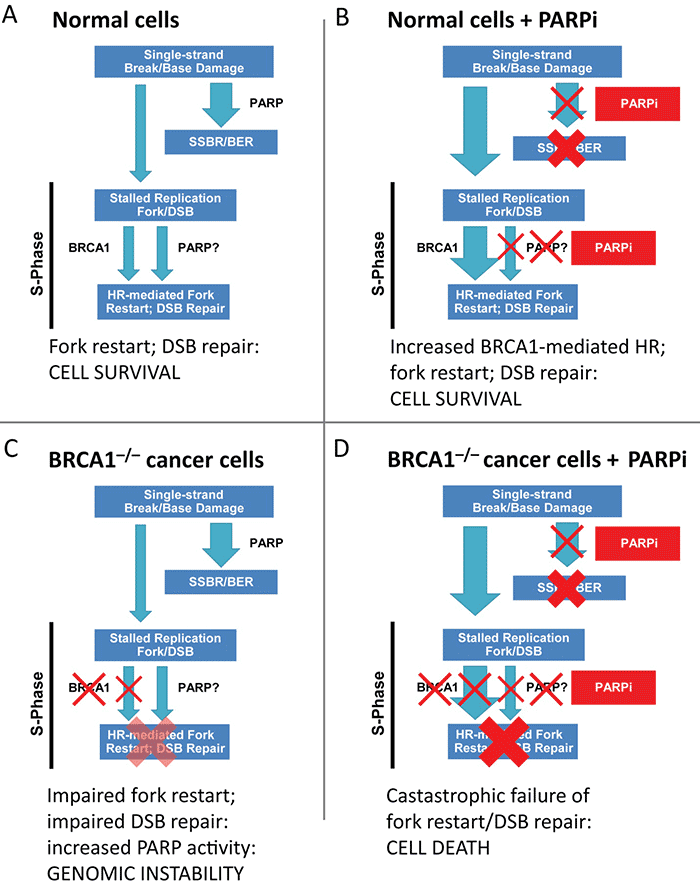
Figure 1. Conditional synthetic lethality of poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) with BRCA1−/− cells. (A) In normal cells, single-strand breaks are repaired efficiently by PARP mediated single-strand break repair (SSBR). The few unrepaired breaks that persist and lead to replication fork arrest and double-strand breaks (DSBs) are efficiently handled by BRCA1-dependent repair. (B) In the setting of PARPi, SSBR is impaired leading to accumulation of unrepaired single-strand breaks. This results in a great increase in the amount of replication fork lesions that must be repaired by a BRCA1-dependent repair pathway. (C) In BRCA1−/− cells, homologous recombination (HR)-mediated repair of stalled replication forks and DSBs is impaired; there may be increased reliance of PARP-dependent mechanisms and increased PARP activity. (D) In BRCA1−/− cells treated with PARPi, an increased burden of unrepaired single-strand breaks enter S-phase. The resulting lesions cannot be repaired, as BRCA-mediated repair is completely disabled, leading to catastrophic failure of replication fork restart, accumulation of unrepaired DNA DSBs, and cell arrest/death. BER, base excision repair. Adapted from Aly A, Ganesan S. BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J Mol Cell Biol. 2011;3(1):66-74 by permission of Oxford University Press. http://www.advanceweb.com/web/AstraZeneca/ClinicalTopicsNewsletterSeries/issue-4-oncology.html
The major limitation of PARP inhibition, however, is the requirement for background BRCA1/2 deficiency and the development of compensatory mutations that invoke nonhomologous end joining (NHEJ) or restore HR.
How can HR be disrupted without BRCA1/2 mutations?
BRCA1/2 is a natural target of interest in the development of antineoplastic drugs. However, BRCA1/2 is not like traditional targets, for example, kinases, the function of which are blocked with small molecule kinase inhibitors, or antibodies to the extracellular domains of receptor tyrosine kinases. Blocking BRCA1/2 function involves disruption of protein-protein interactions, which are not easily druggable targets.
Onxeo has come up with a different strategy – overwhelm the cell with decoy double strand breaks that occupy the BRCA1/2 machinery, thereby rendering it unavailable to repair chromosomal double strand breaks. This is achieved with AsiDNA’s (decoy agonists of DNA repair machinery) comprised of 32 base-pair DNA duplex tethered to cholesterol to facilitate nuclear localization:
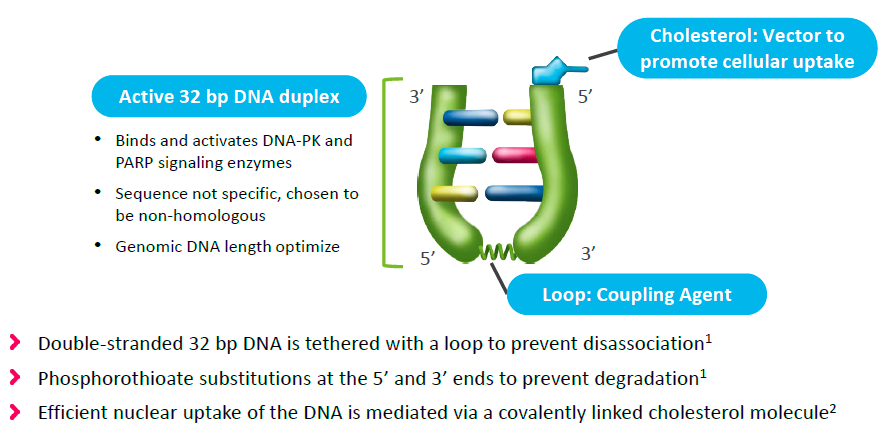
Figure 2. AsiDNA constructs. http://www.onxeo.com/site/wp-content/uploads/2018/01/180102_Onxeo_Presentation_JPM_D4-for-print.pdf
A Phase 1 proof of concept clinical trial of intratumoral injection combined with RT in patients with metastatic melanoma demonstrated a 59% response rate (30% complete response rate), as well as abscopal effects, that is, shrinkage of non-injected lesions outside the field of radiation.
In a mouse xenograft study of triple negative breast cancer (TNBC, synergy between intravenous AsiDNA and PARP inhibition (olaparib, Lynparza)was demonstrated. (Lynparza was recently approved for use in patients with BRCA-mutated Her-2 negative metastatic breast cancer.) In vitro studies in 12 breast cancer cell lines have demonstrated that concomitant administration of olaparib and AsiDNA leads to inhibition of XRCC1 and RAD51/53BP1 repair enzymes required for BER and HR, respectively.
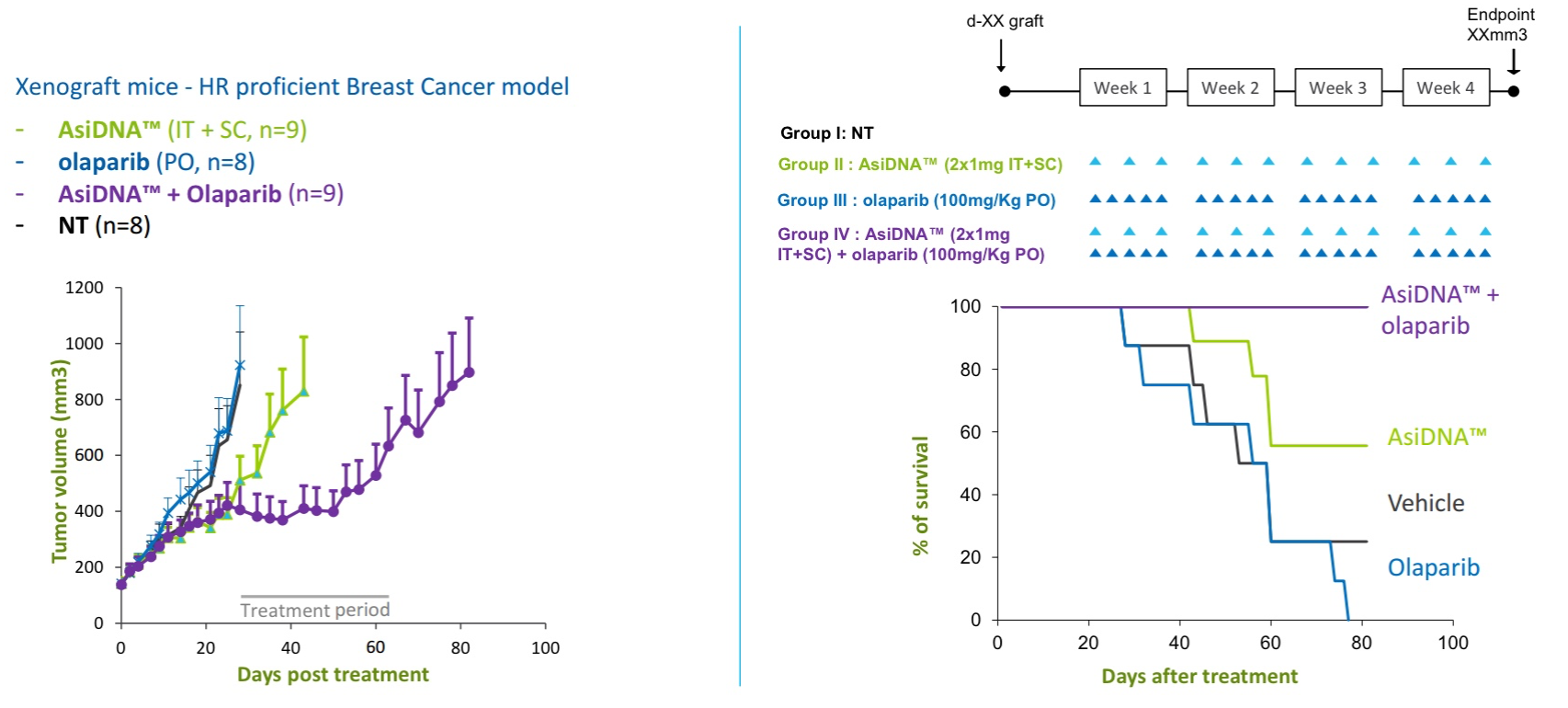
Figure 3. Synergy between intravenous AsiDNA and olaparib in xenograft model of triple negative breast cancer. http://www.onxeo.com/site/wp-content/uploads/2018/01/180102_Onxeo_Presentation_JPM_D4-for-print.pdf
The company is filing an IND to perform clinical studies in combination with PARP inhibitors. Rather than limit use of PARP inhibitors to patients with BRCA1 mutations, the goal is for the AsiDNA to functionally block BRCA1 by occupying the cellular machinery; then, single strand breaks induced by PARP inhibition will fatally overwhelm the cell with double strand breaks that cannot be repaired in time to stop mitotic catastrophe.
Can agents other than PARP inhibitors be combined with AsiDNA to precipitate mitotic catastrophe in wild type BRCA cancer cells?
The histone deacetylase (HDAC) inhibitor, belinostat, has been shown to induce genetic instability by causing single strand breaks in cancer cell lines. This is accomplished by (1) opening chromatin and rendering DNA susceptible to attack by (2) induced reactive oxygen species. These effects are not observed in healthy cell lines.
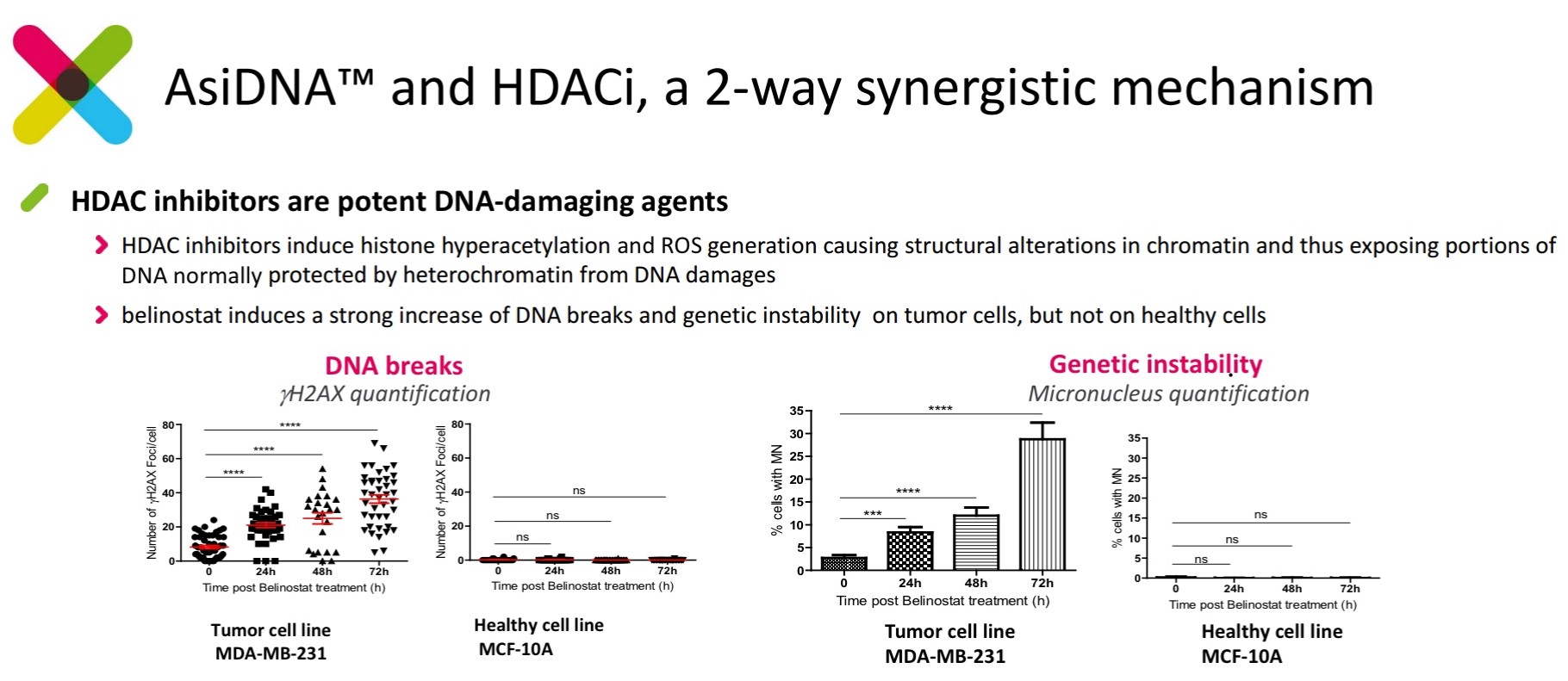
Figure 4. Belinostat (HDAC inhibitor) induces DNA damage and genetic instability in cancer cell lines, but not wild type cells. http://www.onxeo.com/site/wp-content/uploads/2018/01/180102_Onxeo_Presentation_JPM_D4-for-print.pdf
Given that belinostat causes single strand breaks, would combination with AsiDNA produce similar effects seen with PARP inhibition and AsiDNA exposure? Indeed, Significant synergy has been demonstrated when combining AsiDNA and belinostat in cancer cell lines. Importantly, this synergy is not observed in wild type cells.

Figure 5. Synergy of belinostat (HDAC inhibitor) and AsiDNA lethality on tumor cell lines but not healthy cell lines. http://www.onxeo.com/site/wp-content/uploads/2018/01/180102_Onxeo_Presentation_JPM_D4-for-print.pdf
The molecular basis for the synergy is familiar: (1) HDAC inhibitors induce DNA damage (similar to PARP inhibitors), leading to double strand breaks; (2) AsiDNA occupies the HR double strand repair machinery; (3) the HR repair machinery cannot repair chromosomal DNA double strand breaks before mitotic catastrophe occurs.
The company is planning to conduct clinical studies of belinostat and AsiDNA, as well.