

Bristol-Myers Squibb recruited French biotech Oncodesign to help it find new ways of attacking tumors, signing an early-stage agreement in hopes of spotlighting some promising projects.
Key to the collaboration is Oncodesign’s Nanocyclix platform, Bristol-Myers said, through which the company says it can boost the potency and binding power of small-molecule therapies. Oncodesign has a library of more than 5,000 kinase inhibitors stemming from Nanocyclix, targeting cancer and other diseases.
The Oncodesign Approach
Nanokinib is a kinase focused library of proprietary small molecule macrocycles that Oncodesign has created using its Nanocyclix® technology. The current library contains over 5,000 diverse kinase inhibitors that were broadly profiled across the human kinome and in early ADMET (eADMET) models. The compounds typically display low nanomolar potencies against a small number of kinases. Inhibitor leads have been discovered both for established and unexplored kinases. Each compound of the Nanokinib library was individually designed to display good cell penetration and attractive physicochemical properties.
Conceptually, the Nanocyclix® technology enhances a ligand’s potency and selectivity towards its receptor due to conformational pre-organization and tighter binding site recognition. In our experience, exploring different lengths and functionalities of the cyclic linker allow to identify an optimal match between the size and mobility of the binding site and the macrocyclic ligand. Once an initial “good fit” scaffold/linker combination has been identified, the available platform knowledge is applied to further optimize the compounds.
Conventional kinase focused collections are based on extensive chemical modifications of 1 or a small number of so-called “kinase scaffolds”, and as a consequence only have a limited degree of diversity. These “kinase scaffolds” bind to the hinge region of kinase active sites as they mimic ATP which is the natural source of the phosphate group that is used by kinases for the phosphorylation of their substrates.
Oncodesign’s Nanokinib library provides a much higher degree of diversity as it is based on over 50 known and novel “kinase scaffolds” that are combined with a preferred set of over 300 “linkers” (the cyclic bridge in the final molecules that forms the macrocyclic ring). As a consequence, Nanokinib allows to address kinases within the different subfamilies across the kinome, including unexplored kinases for which currently no inhibitors exist.
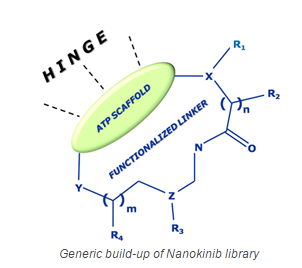
Despite being macrocyclic, the Nanokinib molecules are structurally simple as they contain in general no chiral groups. A high degree of potency and selectivity is achieved within the core macrocyclic structure, within a molecular weight of 300-400. This is in strong contrast to potent and selective “open form” kinase inhibitors, which are heavily functionalized to access remote kinase specificity pockets and consequently have molecular weights close to 600.
Current Designs of Kinases
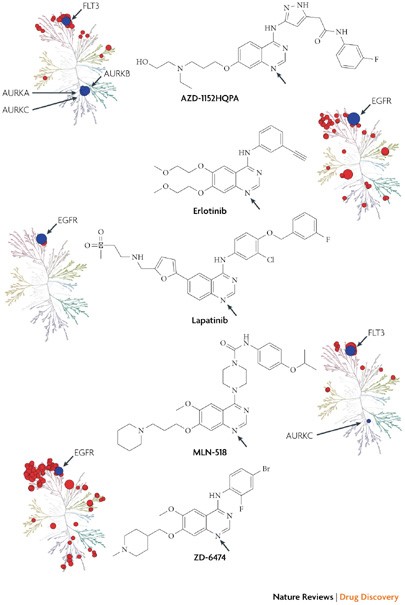
Other kinase inhibitors that are approved and in development target the ATP binding site. The quinazoline ring has frequently been used as a core scaffold to occupy the adenine ring region of the ATP binding site.
The kinase interaction maps for five quinazoline compounds — AZD-1152HQPA, erlotinib (Tarceva; OSI/Genentech/Roche), lapatinib (Tykerb; GlaxoSmithKline), MLN-518 and ZD-6474 — show that compounds based on the quinazoline scaffold can target a range of kinases with varying degrees of selectivity. A superposition of the binding conformation of these inhibitors reveals that the quinazoline is in approximately the same position and forms a key hydrogen bond between the quinazoline N1 (see arrows in the accompanying figures) and the kinase hinge region. Lapatinib is the most selective compound in this set and only interacts with the epidermal growth factor receptor (EGFR) subfamily. Presumably this is due to the ability of lapatinib to recognize an inactive conformation of EGFR47 that is distinct from the classical active conformation recognized by the type I inhibitors erlotinib50 and ZD-6474. Erlotinib and ZD-6474 show considerably broadened kinase interaction profiles.
AZD-1152HQPA52 (Barasertib) and MLN-518 (Tandutinib) are type II inhibitors that are predicted to recognize an inactive conformation in which the activation loop blocks substrate binding, known as the DFG-out conformation. The interaction map reveals that AZD-1152HQPA binds with high affinity to FLT3.
http://www.nature.com/nrd/journal/v7/n5/box/nrd2541_BX1.html