

CANCER RESEARCH UK scientists have found a drug combination that can trigger the self-destruct process in lung cancer cells – paving the way for new treatments, according to research that will be presented at the National Cancer Research Institute (NCRI) Cancer Conference in Liverpool next week. This process is known as apoptosis.
The discovery, although still in its infancy, could essentially revolutionize the way doctors approach cancer. “Igniting the fuse that causes lung cancer cells to self-destruct could pave the way to a completely new treatment approach — and leave healthy cells unharmed,” lead researcher Dr. Henning Walczak, from University College London Cancer Institute.
In the study, the team used lung cancer cells from mice to demonstrate how two drugs, TRAIL and a CDK9 inhibitor, altered the molecular switches of the cancer cells, forcing them to kill themselves Mission Impossible style. Impressively, none of the mice’s healthy cells were negatively affected by the potent drug combination.
TRAIL (TNF-related apoptosis inducing ligand) binds to the Fas Death Receptor triggering the conversion of Caspases 8 and 10, thereby invoking the extrinsic apoptotic program.
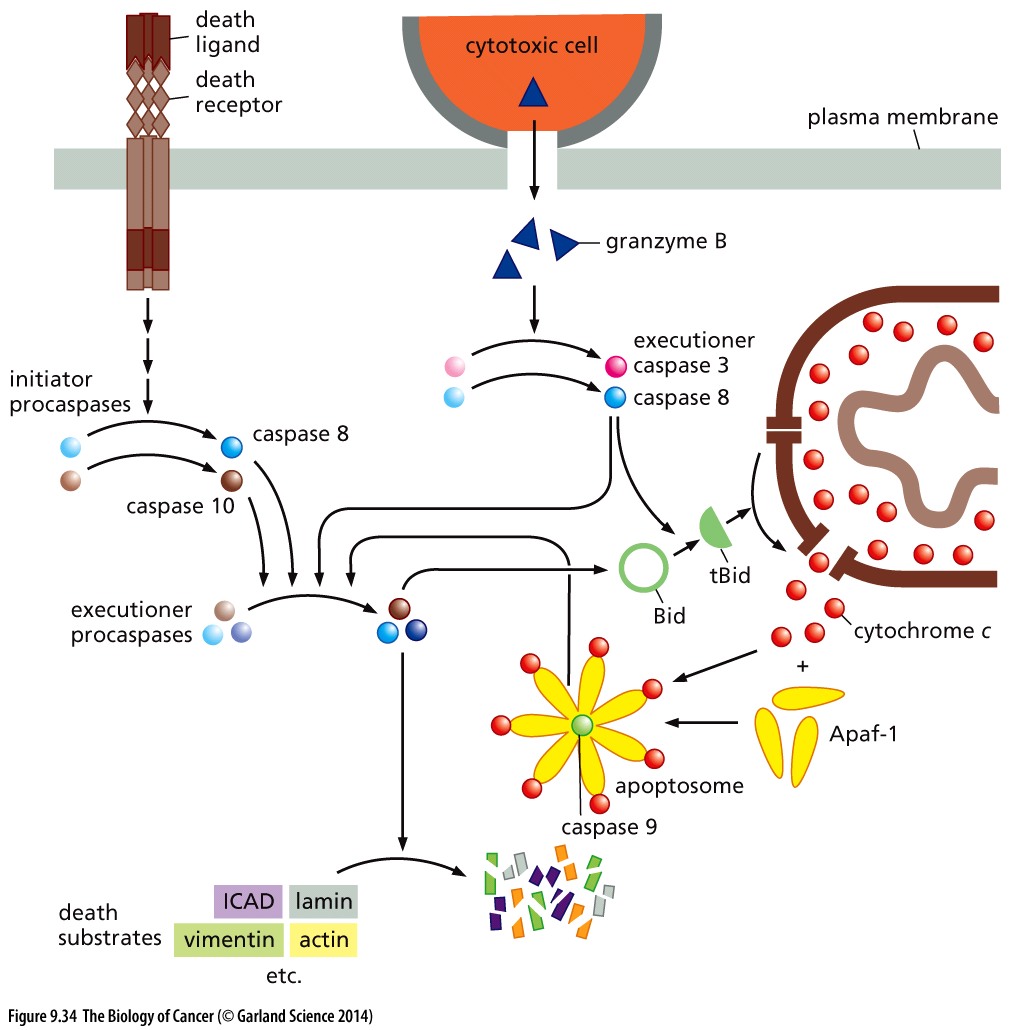
Copyright 2014 from The Biology of Cancer, 2nd Ed. by Weinberg. Reproduced by permission of Garland Science/Taylor & Francis LLC
CDK9 is responsible for phosphorylating Mdm2, a molecule which tags p53 for ubiquitylation and thereby stopping the intrinsic apoptotic program.
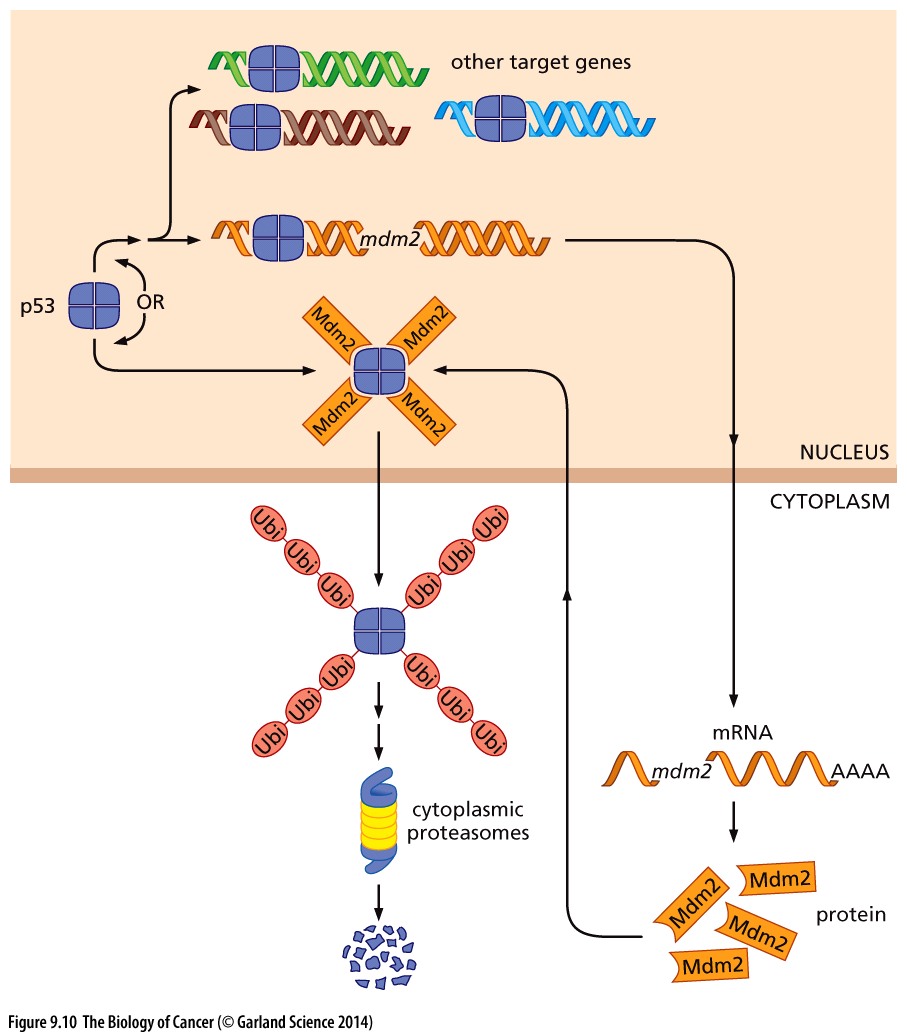
Copyright 2014 from The Biology of Cancer, 2nd Ed. by Weinberg. Reproduced by permission of Garland Science/Taylor & Francis LLC
CDK9 also forms the catalytic core of the positive transcription elongation factor b (P-TEFb). This enzyme is critical for stimulating transcription elongation of most protein coding genes, including key developmental and stimulus-responsive genes, by RNA polymerase II (RNAPII). CDK9 regulates cytokine inducible transcription networks by facilitating promoter recognition of target transcription factors (e.g. TNF-inducible RELA/p65 activation and IL-6-inducible STAT3 signaling). It promotes RNA synthesis in genetic programs for cell growth, differentiation and viral pathogenesis.

http://m.ztopics.com/CDK9/
CDK9 inhibition causes the rapid depletion of short-lived mRNA transcripts and their associated protein products. Since many genes encoding proteins involved in cell growth, proliferation, and survival are characterized by short-lived mRNAs and proteins, the consequences of CDK9 inhibition are expected to include anti-proliferative and pro-apoptotic effects through loss of function at many cellular pathways. Proteins with well-established roles in tumor development and growth that are depleted following CDK9 inhibition include Myc, Cyclin D1, and Mcl-1, the long isoform of which is pro-survival, that is, blocks apoptosis. Cyclin D-1 drives the cell cycle via hypophosphorylation of Rb; myc drives the cell cycle clock by inducing Id pocket proteins, which neutralize pRb and myc also indiced hTERT, thereby immortalizing cancer cells. Blocking CDK9, therefore, removes the inhibition of apoptosis (Mdm2 and Mcl-1), and removes drivers of proliferation (Cyclin D-1 and myc), and immortalization.
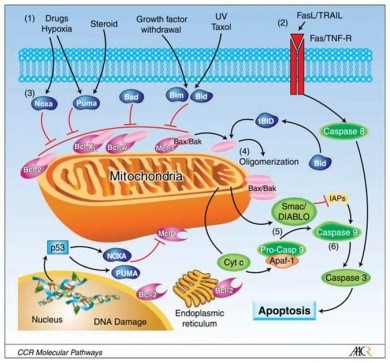
http://www.priaxon.com/content/technology/mcl1-inhibitors/index.php