

It is widely known that brain tumors, in particular glioblastomas and medulloblastomas, affect males more than females. Moreover, given the age of onset and analysis of incidence by age, sex hormones have been ruled-out as the cause.
What is?
A paper from the Washington University in the Journal of Clinical Oncology sheds light on this observation. The group leader, Joshua Rubin, stated that the team knew that sex hormones could not account for the differences in brain tumor risk.
“Male brain tumor risk remains higher throughout life despite major age-linked shifts in sex hormone production in males and females,” he said. “If the sex hormones were causing this effect, we’d see major changes in the relative rates of brain tumors in males and females at puberty. But they don’t happen then or later in life when menopause changes female sex hormone production.”
Rubin used a cell model of glioblastoma to prove it is easier to make male brain cells become tumors. After a series of genetic alterations and exposure to a growth factor, male brain cells became cancerous faster and more often than female brain cells.
In experiments designed to identify the reasons for the differences in the male and female cells, the team evaluated three genes to see if they were naturally less active in male brain cells. The genes they studied — neurofibromin, p53 and RB — normally suppress cell division and cell survival. They are mutated and disabled in many cancers.
The scientists found RB was more likely to be inactivated in male brain cells than in female brain cells. When they disabled the RB protein in female brain cells, the cells were equally susceptible to becoming cancers.
The RB (retinoblastoma) gene is a tumor suppressor located on the short arm of chromosome 13. Germ line mutations in both alleles are associated with familial retinoblastoma and osteosarcoma. Somatic cell gene methylation or loss of heterozygosity of the wild-type allele results in sporadic cases of retinoblastoma.
RB is the ultimate regulator of the cell cycle. As such, it is the final common pathway of all growth signals that lead to cell division. Perturbation of its function has grave consequences with respect to the cellular homeostatic controls and checks and balances on cell division.
Un-phosphorylated RB keeps the cell in G1; hypophosphorylated RB by cyclin D-CDK4/6 ushers in the R-point transition, that critical point late in G1 of the cell cycle when the cell is committed to dividing and is unresponsive to external cues (pro growth versus pro differentiation).
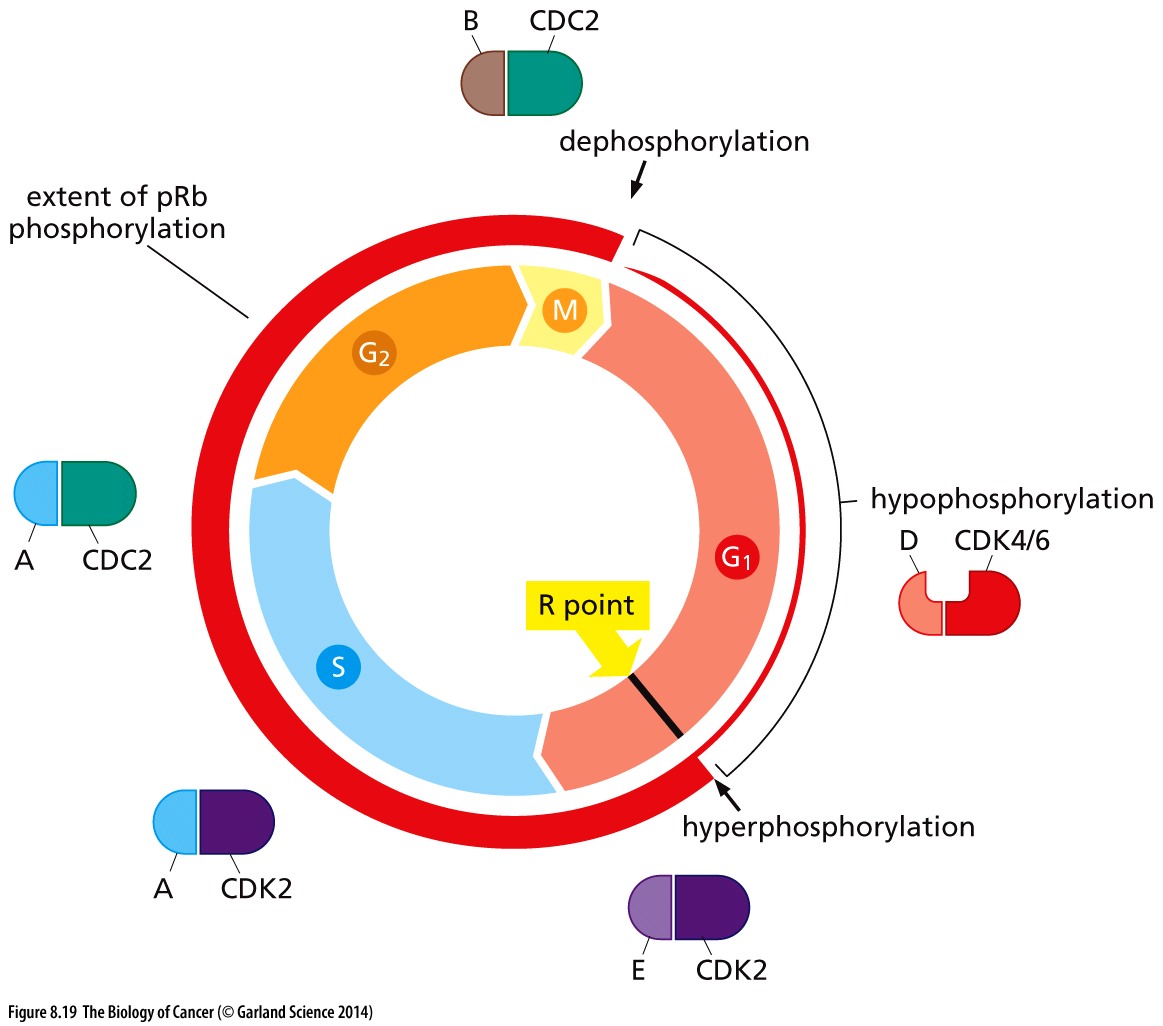
Cell cycle–dependent phosphorylation of pRb. The phosphorylation state of pRb (red circle) is closely coordinated with cell cycle advance. As cells pass through the M/G1 transition, virtually all of the existing phosphate groups are stripped off pRb, leaving it in an unphosphorylated configuration. As cells progress through G1, a single phosphate group is attached as any one of 14 different phosphorylation sites (by cyclin D-CDK4/6 complexes), yielding hypophosphorylated pRb. However, when cells pass through the restriction (R) point, cyclin E–CDK2 complexes phosphorylate pRb on at least 12 more sites, placing it in a hyperphosphorylated state. Throughout the remainder of the cell cycle, the extent of pRb phosphorylation remains constant until cells enter into M phase. Copyright 2014 from The Biology of Cancer, 2nd Ed. by Weinberg. Reproduced by permission of Garland Science/Taylor & Francis LLC
Hyperphosphorylation of RB resulting from cyclin E-CDK2 activity inactivates RB, as does HPV protein E7 and Id proteins that are induced by myc. Inactivated RB allows the cell cycle to continue through mitosis.
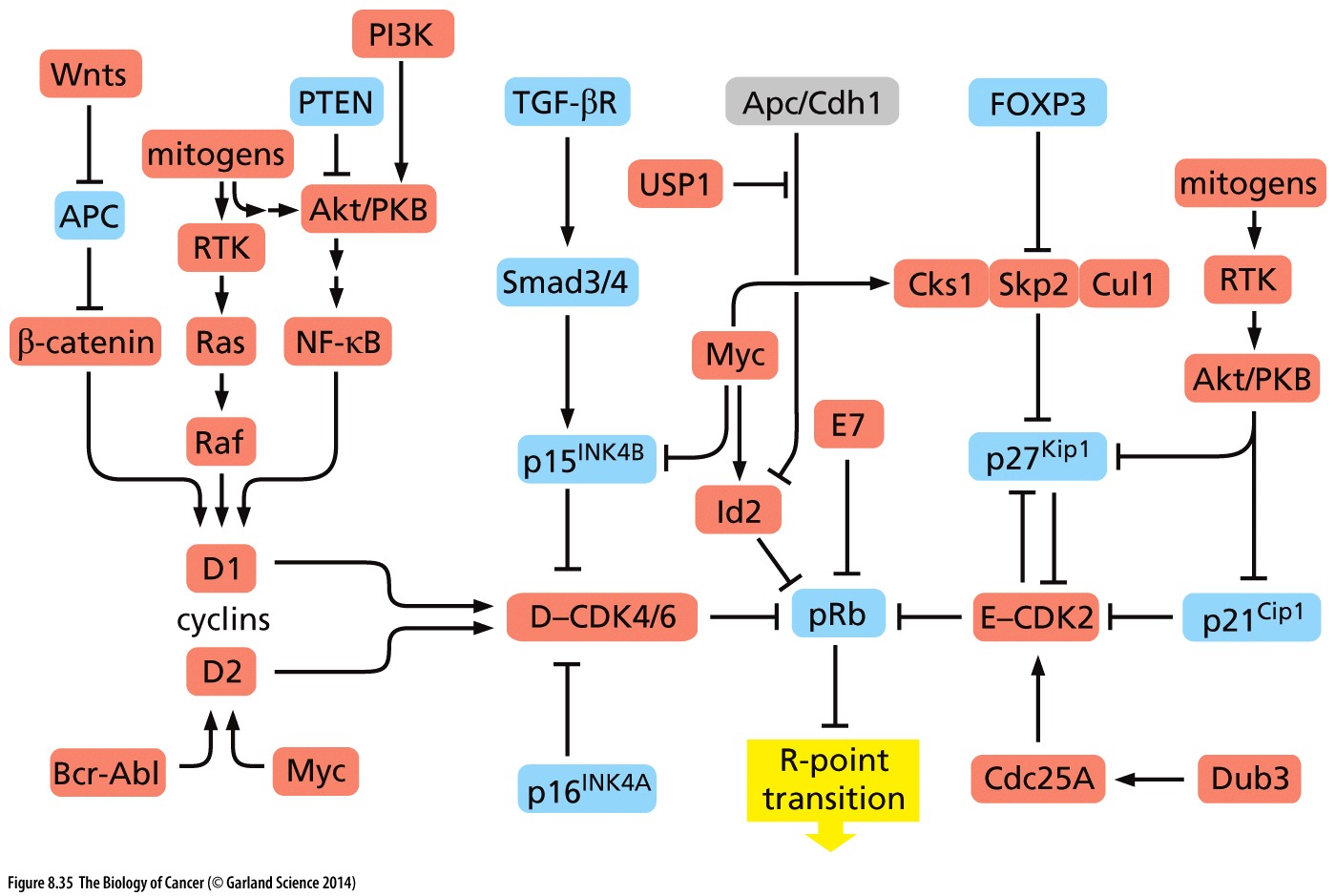
Copyright 2014 from The Biology of Cancer, 2nd Ed. by Weinberg. Reproduced by permission of Garland Science/Taylor & Francis LLC
I want to thank Professor Roberta Moldow for sending me this paper for the blog.